
ЮЯЯ ЯЮПЯ
Я
ЮЮБЯЮЏЮЛЮЕЮЙЮП ЮЁЮБЮМЯЮЏЮДЮЗ ЮКЮБЮЙ ЮЮЙЮКЯЮЛЮБЮП ЮЮЕЮНЯЮЕЯЮПЮЖЮЏЮДЮЗ
Ю ЮБЮИЮПЮЛЯЮГЮПЯ
Я ЮЮГЮКЮПЮЛЯЮГЮПЯ
Я
Ю ЮКЮБЯЮКЮЏЮНЮПЯ ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ ЮБЯЮПЯЮЕЮЛЮЕЮЏ ЯЮЗЮН ЯЯЯЯЮЕЯЮПЯ ЯЮБ ЮБЮЙЯЮЏЮБ ЮИЮНЮЗЯЮЙЮМЯЯЮЗЯЮБЯ ЯЮБЮГЮКЮПЯЮМЮЏЯЯ. Ю ЮЕЯЮЏЯЮПЯ 230000 ЮНЮЮЕЯ ЯЮЕЯЮЙЯЯЯЯЮЕЮЙЯ ЮКЮБЯЮКЮЏЮНЮПЯ ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ ЮЕЮКЯЮЙЮМЮЌЯЮБЮЙ ЯЯЮЙ ЮДЮЙЮБЮГЮЙЮГЮНЯЯЮКЮПЮНЯЮБЮЙ ЮЕЯЮЗЯЮЏЯЯ ЮМЯЮНЮП ЯЯЮЙЯ ЮЮНЯЮМЮЮНЮЕЯ Ю ЮПЮЛЮЙЯЮЕЮЏЮЕЯ ЮЮМЮЕЯЮЙЮКЮЎЯ ЮКЮБЮЙ ЮПЮЙ ЮБЮНЮБЮМЮЕЮНЯЮМЮЕЮНЮПЮЙ ЮИЮЌЮНЮБЯЮПЮЙ ЮЕЮЏЮНЮБЮЙ ЯЮЕЯЮЏ ЯЯЮН 160000. Ю ЮБЯЮЌ ЯЮЗЮН ЯЯЯЮПЮДЮП ЯЯЮЗЮН ЮЯЮЕЯ ЮНЮБ ЯЮПЯ ЮКЮБЯЮКЮЏЮНЮПЯ , ЮЗ ЯЮЕЮНЯЮБЮЕЯЮЎЯ ЮЕЯЮЙЮВЮЏЯЯЮЗ ЯЮБЯЮБЮМЮЮНЮЕЮЙ ЯЯЯЯЮЎ ЮКЮБЮЙ ЮБЮГЮГЮЏЮЖЮЕЮЙ ЯЮП 15%.Т ЮЮЕ ЮМЮЙЮБ ЮЙЯЯЮПЯЮЙЮКЮЎ ЮМЮБЯЮЙЮЌ, ЮП ЮМЮЗ ЮМЮЙЮКЯЮПЮКЯ ЯЯЮБЯЮЙЮКЯЯ ЮКЮБЯЮКЮЏЮНЮПЯ ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ (NSCLC) ЮБЮНЯЮЙЮМЮЕЯЯЯЮЙЮЖЯЯЮБЮН ЯЮБЮН ЮМЮЏЮБ ЮЕЮНЮЙЮБЮЏЮБ ЮНЮПЯЮПЮЛЮПЮГЮЙЮКЮЎ ЮПЮНЯЯЯЮЗЯЮБ ЮКЮБЮЙ ЮЗ ЯЮЗЮМЮЕЮЙЮПЮИЮЕЯЮБЯЮЕЮЏЮБ ЯЯЮП ЮМЮЕЯЮБЯЯЮБЯЮЙЮКЯ ЮКЮБЯЮКЮЏЮНЮП ЮЕЮЏЯЮЕ ЯЮЕЯЮПЯЮЙЯЮМЮЮНЮБ ЮБЯЮПЯЮЕЮЛЮЯЮМЮБЯЮБ ЯЯЯЮН ЮБЯЮПЯЮЌ ЯЮЗ ЯЯ ЮНЮПЮЛЮЙЮКЮЎ ЮЕЯЮЙЮВЮЏЯЯЮЗ ЮКЮБЮЙ ЯЮЗЮН ЮЕЮОЮБЯЯЮЌЮЛЮЙЯЮЗ ЮЙЮКЮБЮНЮПЯЮПЮЙЮЗЯЮЙЮКЮЎЯ ЯЮПЮЙЯЯЮЗЯЮБЯ ЮЖЯЮЎЯ. ЮЮЙЮБ ЯЮЕЮЙЯЮЌ ЮБЯЯ ЮМЮЕЮГЮЌЮЛЮЕЯ ЯЯ ЯЮБЮЙЮПЯЮПЮЙЮЗЮМЮЮНЮЕЯ ЯЮЌЯЮЗЯ ЮЮЮ ЮКЮЛЮЙЮНЮЙЮКЮЯ ЮМЮЕЮЛЮЯЮЕЯ ЮКЮБЮИЮЙЮЯЯЯЮБЮН ЯЮЗ ЯЮЗЮМЮЕЮЙЮПЮИЮЕЯЮБЯЮЕЮЏЮБ ЮМЮЕ ЮДЮЙЯЮЛЮЯЮБ ЮВЮБЯЮЙЯЮМЮЮНЮЗ ЯЯЮЗЮН ЯЮЛЮБЯЮЏЮНЮБ ЯЯ standard of care ЯЯЮЗ ЮИЮЕЯЮБЯЮЕЮЏЮБ ЯЮПЯ ЮМЮЕЯЮБЯЯЮБЯЮЙЮКЮПЯ ЮМЮЗ ЮМЮЙЮКЯЮПЮКЯ ЯЯЮБЯЮЙЮКЮПЯ ЮКЮБЯЮКЮЏЮНЮПЯ ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ ЮМЮЕ ЯЮПЯЮПЯЯЮЌ ЮБЮНЯЮБЯЯЮКЯЮЙЯЮЗЯ 20 ЮМЮЕ 30 % ЮКЮБЮЙ ЮМЮЯЮЗ ЮЕЯЮЙЮВЮЏЯЯЮЗ 8 ЮМЮЕ 11 ЮМЮЎЮНЮЕЯ. ЮЯ ЯЮЎ ЮЗ ЮИЮЕЯЮБЯЮЕЯ ЯЮЙЮКЮЎ ЯЯЮПЯЮЮГЮГЮЙЯЮЗ ЯЮБЮЏЮНЮЕЯЮБЮЙ ЮНЮБ ЮЯЮЕЮЙ ЯЯЮЌЯЮЕЮЙ ЯЮЕ ЮЮНЮБ plateau ЮБЯЮПЯЮЕЮЛЮЕЯЮМЮБЯЮЙЮКЯЯЮЗЯЮБЯ ЮКЮБЮЙ ЮБЯ ЯЯ ЮПЮДЮЎЮГЮЗЯЮЕ ЯЮЕ ЮНЮЮЕЯ ЮЕЮОЮЕЮЛЮЏЮОЮЕЮЙЯ ЯЯЮЗ ЮДЮЙЮБЯЮЕЮЏЯЮЗЯЮЗ ЮБЯ ЯЯЮН ЯЯЮН ЮБЯЮИЮЕЮНЯЮН.
Ю ЯЯЯЯЮБЯЮЕЯ ЮМЮЕЮЛЮЯЮЕЯ ЯЯЮЗЮН ЮБЮНЯЮЙЮМЮЕЯЯЯЮЙЯЮЗ ЯЮПЯ ЮМЮЕЯЮБЯЯЮБЯЮЙЮКЮПЯ ЮМЮЗ ЮМЮЙЮКЯЮПЮКЯ ЯЯЮБЯЮЙЮКЮПЯ ЮКЮБЯЮКЮЏЮНЮПЯ ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ (NSCLC) ЮПЮДЮЎЮГЮЗЯЮБЮН ЯЯЮЗ ЮДЮЙЮБЯЮЏЯЯЯЯЮЗ ЯЯЮЙ ЮП NSCLC ЮДЮЕЮН ЮБЯЮПЯЮЕЮЛЮЕЮЏ ЮБЯЮПЮКЮЛЮЕЮЙЯЯЮЙЮКЮЌ ЮМЮЏЮБ ЮНЮПЯЮПЮЛЮПЮГЮЙЮКЮЎ ЮПЮНЯЯЯЮЗЯЮБ, ЮБЮЛЮЛЮЌ ЮДЮЙЮБЯЮПЯЮЕЯЮЙЮКЮЌ ЮНЮЕЮПЯЮЛЮЌЯЮМЮБЯЮБ ЮКЮБЮИЮПЮДЮЗЮГЮПЯЮМЮЕЮНЮБ ЮБЯЯ ЮДЮЙЮБЯЮПЯЮЕЯЮЙЮКЮЌ ЮМЮПЯЮЙЮБЮКЮЌТ ЮМЮПЮНЮПЯЮЌЯЮЙЮБ, ЮДЮЙЮБЯЮПЯЮЕЯЮЙЮКЮЎ ЮВЮЙЮПЮЛЮПЮГЮЙЮКЮЎ ЯЯ ЮМЯЮЕЯЮЙЯЮПЯЮЌ ЮКЮБЮЙ ЮКЮБЯт ЮЕЯЮЮКЯЮБЯЮЗ ЯЯЮЎЮЖЮПЯ ЮН ЮДЮЙЮБЯЮПЯЮЕЯЮЙЮКЮЎ ЮБЮНЯЮЙЮМЮЕЯЯЯЮЙЯЮЗ (ЮЕЮЙЮКЯЮНЮБ 1). Ю Lynch ЮКЮБЮЙ ЯЯ ЮНЮЕЯЮГЮЌЯЮЕЯ ЮКЮБЮЙ ЮП Paez ЮКЮБЮЙ ЯЯ ЮНЮЕЯЮГЮЌЯЮЕЯ ЮЎЯЮБЮН ЮПЮЙ ЯЯЯЯЮПЮЙ ЯЮПЯ ЯЮЕЯЮЙЮЮГЯЮБЯЮБЮН ЮЮНЮБ Я ЯЮПЯЯЮНЮПЮЛЮП ЮБЯЮИЮЕЮНЯЮН ЮМЮЕ ЮМЮЗ ЮМЮЙЮКЯЮПЮКЯ ЯЯЮБЯЮЙЮКЯ ЮКЮБЯЮКЮЏЮНЮП ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ ЯЮПЯ ЮЕЮМЯЮБЮНЮЏЮЖЮПЯ ЮН ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЯЮПЯ Т EGFR ЮГЮПЮНЮЙЮДЮЏЮПЯ ЮКЮБЮЙ ЮПЮЙ ЮПЯЮПЮЏЮПЮЙ ЮБЮНЯЮБЯЮПЮКЯЮЏЮИЮЗЮКЮБЮН ЯЮЕ ЮИЮЕЯЮБЯЮЕЮЏЮБ ЮМЮЕ ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЯЯ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ ЯЮЗЯ ЯЯ ЯЮПЯЮЙЮНЮЙЮКЮЎЯ ЮКЮЙЮНЮЌЯЮЗЯ (TKIs). Ю ЮБЮНЮБЮКЮЌЮЛЯ ЯЮЗ ЮБЯ ЯЮЎ ЮБЯЮПЯЮЮЛЮЕЯЮЕ ЮДЮЙЮБ ЯЮБЮНЯЯЯ ЯЮЗ ЮИЮЕЯЮБЯЮЕЯ ЯЮЙЮКЮЎ ЯЯЮПЯЮЮГЮГЮЙЯЮЗ ЯЮПЯ ЮМЮЗ ЮМЮЙЮКЯЮПЮКЯ ЯЯЮБЯЮЙЮКЮПЯ ЮКЮБЯЮКЮЏЮНЮП ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ ЮКЮБЮЙ ЮЗ ЮНЮЮБ ЮКЮБЯЮЕЯЮИЯ ЮНЯЮЗ ЮЕЮЏЮНЮБЮЙ ЯЮЛЮЮПЮН ЮМЮЙЮБ ЯЮЙЮП ЮЕЮОЮБЯЮПЮМЮЙЮКЮЕЯ ЮМЮЮНЮЗ ЯЯЮПЯЮЮГЮГЮЙЯЮЗ ЮВЮБЯЮЙЯЮМЮЮНЮЗ ЯЯЮБ ЮЕЮНЮЕЯЮГЮПЯЮПЮЙЮЗЮМЮЮНЮБ ЮМЮПЯЮЙЮБЮКЮЌ ЮМЮПЮНЮПЯЮЌЯЮЙЮБ ЯЮПЯ ЯЮГЮКЮПЯ . ЮЮЙЮБ ЯЯЮПЯЮЮГЮГЮЙЯЮЗ ЯЮПЯ ЮДЮЕЮН ЮБЯЮПЯЮЌ ЮМЯЮНЮП ЯЮЙЯ ЮИЮЕЯЮБЯЮЕЮЏЮЕЯ ЯЮПЯ ЯЯЮПЯЮЕЯЮПЯ ЮН EGFR Я ЯЮПЮДЮПЯЮЕЮЏЯ ЯЮЕ ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЏЮНЯЮМЮБ ЯЮНЮЕЯЮМЮПЮНЮБ ЮМЮЕ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ, ЮБЮЛЮЛЮЌ ЮКЮБЮЙ ЯЮЕ ALK ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЯЮНЮЕЯЮМЮПЮНЮБ ЮКЮБЮЙ ЯЮЙЮП ЯЯЯЯЯЮБЯЮБ ЯЮЕ ЯЮЕЯЮЙЯЯЯЯЮЕЮЙЯ ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЏЮНЯЮМЮБ ЯЮНЮЕЯЮМЮПЮНЮБ ЯЮПЯ ЮЕЮКЯЯЮЌЮЖЮПЯ ЮН ROS1 ЮКЮБЮЙ BRAF ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ.
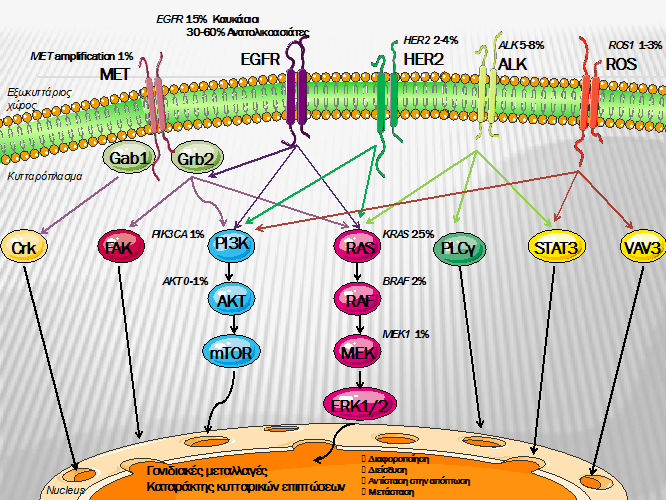
ЮЮЙЮКЯЮНЮБ 1: ЮЯ ЯЯЮБЯЮЙЮКЮЌ ЮМЮПЮНЮПЯЮЌЯЮЙЮБ ЯЯЮП ЮМЮЗ ЮМЮЙЮКЯЮПЮКЯ ЯЯЮБЯЮЙЮКЯ ЮКЮБЯЮКЮЏЮНЮП ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ ЮКЮБЮЙ ЮЗ ЯЯ ЯЮНЯЯЮЗЯЮБ ЯЯЮН ЮПЮДЮЗЮГЯЮН ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЯЮН ЯЯЮП ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЏЮНЯЮМЮБ ЯЮНЮЕЯЮМЮПЮНЮБ
EGFR ЮМЮПЮНЮПЯЮЌЯЮЙ ЯЯЮПЮН ЮКЮБЯЮКЮЏЮНЮП ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ
Т ЮЄЮП EGFR ЮЕЮЏЮНЮБЮЙ ЮМЮЮЛЮПЯ ЯЮЗЯ ЮПЮЙЮКЮПЮГЮЮНЮЙЮЕЮБЯ ЯЯЮН ErbB (ЮЎ HER) Я ЯЮПЮДЮПЯЮЯЮН ЯЮЗЯ ЯЯ ЯЮПЯЮЙЮНЮЙЮКЮЎЯ ЮКЮЙЮНЮЌЯЮЗЯ ЮКЮБЮЙ ЮБЯЮПЯЮЕЮЛЮЕЮЏ ЮКЮБЯЮБЮЛЯ ЯЮЙЮКЯ ЮКЯЮЏЮКЮП ЯЯЮЗЮН ЮЕЮНЮЕЯЮГЮПЯЮПЮЏЮЗЯЮЗ ЮКЯ ЯЯЮБЯЮЙЮКЯЮН ЮМЮПЯЮЙЮБЮКЯЮН ЮМЮПЮНЮПЯЮБЯЮЙЯЮН ЯЯЯЯ ЯЮП RAS-RAF-MEK ЮМЮПЮНЮПЯЮЌЯЮЙ ЮКЮБЮЙ ЯЮП PI3K-AKT-mTOR ЮМЮПЮНЮПЯЮЌЯЮЙ, ЮБЮЛЮЛЮЌ ЮКЮБЮЙ ЮДЯЮНЮБЯЮБЮЙ ЮНЮБ ЮЕЯЮЗЯЮЕЮБЯЯЮПЯЮН ЮКЮБЮЙ ЮБЯЯ ЮЌЮЛЮЛЮПЯ Я Я ЯЮПЮДЮПЯЮЕЮЏЯ ЯЯ ЯЮПЯЮЙЮНЮЙЮКЮЎЯ ЮКЮЙЮНЮЌЯЮЗЯ ЯЯЯЯ ЮП insulin-like growth factor Я ЯЮПЮДЮПЯЮЮБЯ (IGF1-R) ЮКЮБЮЙ ЮЮЮЄ. ЮЄЮБ ЮМЮПЮНЮПЯЮЌЯЮЙЮБ ЮБЯ ЯЮЌ ЮЕЯЮЗЯЮЕЮЌЮЖЮПЯ ЮН ЯЮЗЮН ЮКЯ ЯЯЮБЯЮЙЮКЮЎ ЮДЮЙЮБЯЮПЯЮПЯЮПЮЏЮЗЯЮЗ ЯЮПЯ ЮКЮБЯЮКЮЙЮНЮЙЮКЮПЯ ЮКЯ ЯЯЮЌЯЮПЯ , ЯЮЗЮН ЯЮПЯЮЙЮКЮЎ ЮЕЯЮЮКЯЮБЯЮЗ, ЯЮЗ ЮМЮЕЯЮБЯЯЮБЯЮЙЮКЮЎ ЯЮПЯ ЮЙЮКЮБЮНЯЯЮЗЯЮБ, ЯЮЗЮН ЮБЮНЯЮЏЯЯЮБЯЮЗ ЯЯЮЗЮН ЮБЯЯЯЯЯЯЮЗ ЮКЮБЮЙ ЯЮЗЮН ЮБЮГЮГЮЕЮЙЮПЮГЮЮНЮЕЯЮЗ. ЮЮЙ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЯЮЙЮП ЯЯ ЯЮНЮЌ ЮЛЮБЮМЮВЮЌЮНЮПЯ ЮН ЯЯЯЮБ ЮМЮЕ ЮЮЛЮЛЮЕЮЙЯЮЗ ЯЮПЯ ЮЕЮОЯЮНЮЏЮПЯ 19 (45%) ЮКЮБЮЙ ЮБЮНЯЮЙЮКЮБЯЮЌЯЯЮБЯЮЗ ЯЮПЯ L858R ЯЮПЯ ЮЕЮОЯЮНЮЏЮПЯ 21 (40-45 %) ЮКЮБЮЙ ЯЮЙЮП ЯЯЮЌЮНЮЙЮБ ЮМЮЕ ЮБЮНЯЮЙЮКЮБЯЮЌЯЯЮБЯЮЗ ЮНЮПЯ ЮКЮЛЮЕЮПЯЮЙЮДЮЏЯЮН ЯЯЮП ЮЕЮОЯЮНЮЙЮП 18 ЮКЮБЮЙ ЯЯЮПЯЮИЮЎЮКЮЗ ЯЯЮП ЮЕЮОЯЮНЮЙЮП 20 ЯЮЕ ЯЮПЯЮПЯЯЯ 5 %. ЮЮЙ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЮЕЮЏЮНЮБЮЙ ЯЮБЯЮПЯЯЮЕЯ ЯЯЮП 15 % ЯЯЮН ЮВЯЯЮЕЮЙЯЮН ЮБЮМЮЕЯЮЙЮКЮЌЮНЯЮН ЮКЮБЮЙ ЮДЯ ЯЮЙЮКЮПЮЕЯ ЯЯЯЮБЮЏЯЮН ЮМЮЕ ЮКЮБЯЮКЮЏЮНЮП ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ, 30 ЮМЮЕ 50 % ЮБЮНЮБЯЮПЮЛЮЙЮКЯЮН ЮБЯЮЙЮБЯЯЮН, ЮЕЮНЯ ЯЮП ЯЮПЯЮПЯЯЯ ЮОЮЕЯЮЕЯЮНЮЌЮЕЮЙ ЯЮП 50 % ЯЯЮН ЮБЯЮЙЮБЯЯЮН ЯЮПЯ ЮДЮЕЮН ЮКЮЌЯЮНЮЙЯЮБЮН ЯЮПЯЮ ЮМЮЕ ЮЙЯЯЮПЮЛЮПЮГЮЙЮКЮЎ ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЙЮНЯЮМЮБЯЮПЯ.
Ю ЮБЮНЮБЮГЮНЯЯЮЙЯЮЗ ЯЮПЯ ЮВЮБЯЮЙЮКЮПЯ ЯЯЮЛЮПЯ ЯЯЮН ЯЯЮПЯЮЕЯЮПЯ ЯЯЮН ЮИЮЕЯЮБЯЮЕЮЙЯЮН ЯЮЕ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЏЮНЯЮМЮБ ЯЮНЮЕЯЮМЮПЮНЮБ ЮКЮБЮЙ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЮЕЯЮЙЮВЮЕЮВЮБЮЙЯЮИЮЗЮКЮЕ ЮБЯЯ ЯЮЗ ЮМЮЕЮЛЮЯЮЗ IPASS (Iressa Pan-Asia Study), ЮМЮЙЮБ ЯЯ ЯЮБЮЙЮПЯЮПЮЙЮЗЮМЮЮНЮЗ ЯЮЌЯЮЗЯ ЮЮЮ ЮКЮЛЮЙЮНЮЙЮКЮЎ ЮМЮЕЮЛЮЯЮЗ ЯЮПЯ ЮЕЮЏЯЮЕ ЯЯ ЯЯЯЯЮП ЯЮЗЮН ЮЕЮКЯЮЏЮМЮЗЯЮЗ ЯЮЗЯ ЮБЯЮПЯЮЕЮЛЮЕЯЮМЮБЯЮЙЮКЯЯЮЗЯЮБЯ ЯЮПЯ gefitinib ЯЮЕ ЯЯЮГЮКЯЮЙЯЮЗ ЮМЮЕ carboplatin ЮКЮБЮЙ paclitaxel ЯЯЮЗЮН ЯЯЯЯЮЗ ЮГЯЮБЮМЮМЮЎ ЮИЮЕЯЮБЯЮЕЮЏЮБЯ ЯЮЕ ЮЕЯЮЙЮЛЮЕЮГЮМЮЮНЮПЯ Я ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЮЕ ЮЙЯЯЮПЮЛЮПЮГЮЙЮКЮЎ ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЙЮНЯЮМЮБЯЮПЯ, ЮБЯЮЙЮБЯЮЙЮКЮЎ ЮЕЮИЮНЯЯЮЗЯЮБ ЯЮПЯ ЮДЮЕЮН ЮЕЮЏЯЮБЮН ЮКЮБЯЮНЮЏЯЮЕЮЙ ЯЮПЯЮ ЮЎ ЮЎЯЮБЮН ЮЕЮЛЮБЯЯЮЕЮЏЯ ЮКЮБЯЮНЮЙЯЯЮЯ. Ю ЮМЮЕЮЛЮЯЮЗ IPASS ЮБЯЮПЯЮЮЛЮЕЯЮЕ ЯЮЗЮН ЯЯЯЯЮЗ ЮМЮЕЮЛЮЯЮЗ ЯЮПЯ ЯЮБЮНЮЯЯЯЮЕ ЯЮЗЮН Я ЯЮЕЯЮПЯЮЎ ЯЮПЯ gefitinib, ЮЕЮНЯЯ ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЯ ЮБЮНЮБЯЯЮПЮЛЮЮБ ЯЮПЯ EGFR, ЮЮНЮБЮНЯЮЙ ЯЮЗЯ ЮМЮЯЯЮЙ ЯЯЯЯЮЕЮЙЮНЮПЯ ЮКЮБЮИЮЙЮЕЯЯЮМЮЮНЮЗ ЮИЮЕЯЮБЯЮЕЮЏЮБ ЮВЮБЯЮЙЯЮМЮЮНЮЗ ЯЯЮЗЮН ЯЮЛЮБЯЮЏЮНЮБ ЯЮЕ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЮЕ ЮКЮБЯЮКЮЏЮНЮП ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ ЮКЮБЮЙ ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЯЮПЯ EGFR. Ю Я ЯЮЕЯЮПЯЮЎ ЮБЯ ЯЮЎ ЮЕЮКЯЯЮЌЯЯЮЗЮКЮЕ ЮМЮЕ 71,2% ЯЮПЯЮПЯЯЯ ЮБЮНЯЮБЯЮПЮКЯЮЏЯЮЗЯ (response rate / RR) ЮКЮБЮЙ 52 % ЮМЮЕЮЏЯЯЮЗ ЯЮПЯ ЯЯЮЕЯЮЙЮКЮПЯ ЮКЮЙЮНЮДЯЮНЮПЯ Я ЯЮПЯЯЮПЯЮЎЯ (progression-free survival / PFS) ЯЯ ЮГЮКЯЮЙЮНЯЮМЮЕЮНЮП ЮМЮЕ ЯЮЗ ЯЮЗЮМЮЕЮЙЮПЮИЮЕЯЮБЯЮЕЮЏЮБ (p<0.001) (Ю ЮЏЮНЮБЮКЮБЯ 1). ЮЮНЯЮЏЮИЮЕЯЮБ, ЮБЯЮИЮЕЮНЮЕЮЏЯ ЯЮПЯ ЮДЮЕЮН ЮЕЮЏЯЮБЮН ЮМЮЕЯЮЌЮЛЮЛЮБЮОЮЗ ЯЮПЯ EGFR ЮКЮБЮЙ ЮЮЛЮБЮВЮБЮН gefitinib ЮЕЮМЯЮЌЮНЮЙЯЮБЮН ЯЮЕЮЙЯЯЯЮЕЯЮБ ЯЮПЯЮПЯЯЮЌ ЮБЮНЯЮБЯЯЮКЯЮЙЯЮЗЯ (RR) (1.1% ЯЯЮПЯ 23.5% ЮБЮНЯЮЏЯЯЮПЮЙЯЮБ) ЮКЮБЮЙ ЮМЮЙЮКЯЯЯЮЕЯЮП ЮДЮЙЮЌЯЯЮЗЮМЮБ ЮЕЮЛЮЕЯЮИЮЕЯЮП ЮНЯЯЮПЯ (PFS) (HR 2.85, p<0.001; ЯЮЏЮНЮБЮКЮБЯ 1). ЮЃЯ ЮНЮЕЯЯЯ, ЮЕЮЏЮНЮБЮЙ ЮБЮНЮБЯЮПЯЮЕЮЛЮЕЯЮМЮБЯЮЙЮКЯ ЮНЮБ ЮБЮНЯЮЙЮМЮЕЯЯЯЮЏЮЖЮЕЮЙЯ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЮЕ EGFR ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЯЯ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ ЯЯЯЮЏЯ ЮНЮБ ЮЯЮЕЮЙ ЯЯЮПЮЗЮГЮЗЮИЮЕЮЏ ЮМЮПЯЮЙЮБЮКЯЯ ЮЮЛЮЕЮГЯЮПЯ ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЯЮН ЮКЮБЮЙ ЮИЮБ ЯЯЮЯЮЕЮЙ ЯЮЛЮПЮЙ ЮПЮЙ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЮЕ ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЏЮНЯЮМЮБ ЯЮНЮЕЯЮМЮПЮНЮБ ЮНЮБ ЮЕЮЛЮЮГЯЮЕЯЮБЮЙ ЯЮП ЮМЮПЯЮЙЮБЮКЯ ЯЮПЯ Я ЯЯЮПЯЮЏЮЛ.
Ю ЮЏЮНЮБЮКЮБЯ 1. ЮЮЛЮЙЮНЮЙЮКЮЯ ЮМЮЕЮЛЮЯЮЕЯТ ЮМЮЗ ЮМЮЙЮКЯЮПЮКЯ ЯЯЮБЯЮЙЮКЮПЯ ЮКЮБЯЮКЮЏЮНЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ ЮМЮЕ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ |
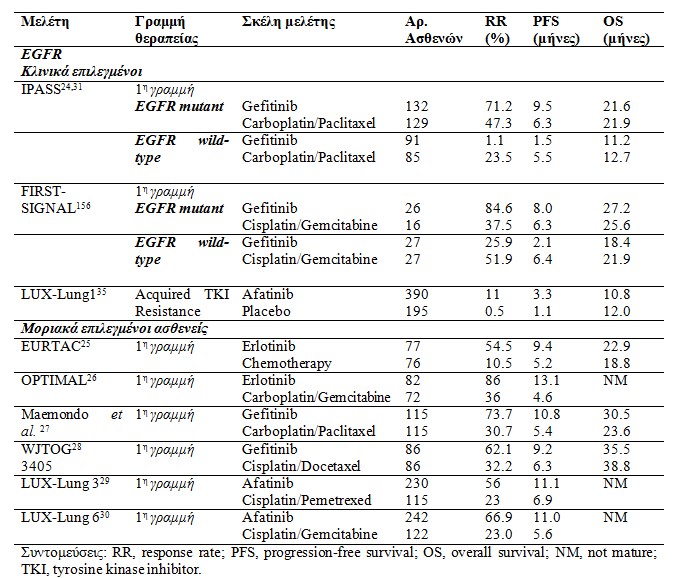
ЮЄЮП ЯЯЮЕЮЛЮПЯ ЮБЯЯ ЯЮЗ ЯЯЮЎЯЮЗ ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЯЮН ЮБЮНЮБЯЯЮПЮЛЮЯЮН ЯЮЕ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЮЕ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЮЕЯЮЙЮВЮЕЮВЮБЮЙЯЮИЮЗЮКЮЕ ЮБЯЯ ЮЮОЮЙ ЯЯ ЯЮБЮЙЮПЯЮПЮЙЮЗЮМЮЮНЮЕЯ ЮМЮЕЮЛЮЯЮЕЯ ЯЮЌЯЮЗЯ ЮЮЮ ЯЮПЯ ЮДЮЙЮЕЯЮЕЯЮНЮЗЯЮБЮН ЯЮП ЯЯЮЛЮП ЯЯЮН gefitinib, erlotinib ЮКЮБЮЙ afatinib. ЮЃЯЮЙЯ ЮМЮЕЮЛЮЯЮЕЯ ЮБЯ ЯЮЯ ЯЮБ ЯЮПЯЮПЯЯЮЌ ЮБЮНЯЮБЯЯЮКЯЮЙЯЮЗЯ (RRs) ЮКЯ ЮМЮБЮЏЮНЮПЮНЯЮБЮН ЮБЯЯ 55 ЮЯЯ 86% ЮКЮБЮЙ ЯЯ ЯЮЕЯЮЏЮЖЮПЮНЯЮБЮН ЮМЮЕ ЮБЮОЮЙЮПЯЮЗЮМЮЕЮЏЯЯЮП ЮДЮЙЮЌЯЯЮЗЮМЮБ PFS (Progression Free Survival) (ЯЮЏЮНЮБЮКЮБЯ 1). Т Ю ЯЮЯЮЕЮЏ ЮНЮБ ЮБЮНЮБЯЮЕЯЮИЮЕЮЏ ЯЯЮЙ ЯЯЯЮП ЮЗ ЮМЮЕЮЛЮЯЮЗ IPASS, ЯЯЮП ЮКЮБЮЙ ЮПЮЙ ЮМЮЕЮЛЮЯЮЕЯ ЮБЯЯ ЯЮПЮН Rossell (EURTAC), ЯЮПЮН Maemondo ЮКЮБЮЙ Mitsudomi (WJTOG 3405) ЮДЮЕЮН ЮБЮНЮЮДЮЕЮЙЮОЮБЮН ЯЯЮБЯЮЙЯЯЮЙЮКЮЌ ЯЮЗЮМЮБЮНЯЮЙЮКЯ ЯЯЮЕЮЛЮПЯ ЮГЮЙЮБ ЯЮПЯ Я ЮБЯЮИЮЕЮНЮЕЮЏЯ ЯЮПЯ ЮЮЛЮБЮВЮБЮН EGFR ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЯЯ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ ЮКЮЙ ЮБЯ ЯЯ ЮДЮЙЯЯЮЙ ЮП ЯЯЮЕЮДЮЙЮБЯЮМЯЯ ЯЯЮН ЮМЮЕЮЛЮЕЯЯЮН ЯЯЮПЮЮВЮЛЮЕЯЮЕТ crossover ЯЯЮН ЮБЯЮИЮЕЮНЯЮН ЯЮПЯ ЮЮЛЮБЮВЮБЮН ЯЮЗЮМЮЕЮЙЮПЮИЮЕЯЮБЯЮЕЮЏЮБ, ЯЯЯЮЕ ЯЮЕ ЯЮЕЯЮЏЯЯЯЯЮЗ Я ЯЮПЯЯЮПЯЮЎЯ ЮНЮБ ЮЛЮЌЮВЮПЯ ЮН ЮКЮЙ ЮБЯ ЯЮПЮЏ EGFR TKIs. ЮЄЮБ ЯЮПЯЮПЯЯЮЌ ЯЯЮН crossover ЮЎЯЮБЮН ЯЮБЯЯЮМЮПЮЙЮБ. 73% ЮГЮЙЮБ ЯЮЗ ЮМЮЕЮЛЮЯЮЗ EURTAC, 95% ЯЯЮЗ ЮМЮЕЮЛЮЯЮЗ ЮБЯЯ ЯЮПЮН Maemondo ЮКЮБЮЙ ЯЯЮН ЯЯ ЮНЮЕЯЮГЮБЯЯЮН ЮКЮБЮЙ 91% ЯЯЮЗ ЮМЮЕЮЛЮЯЮЗ WJTOG.
ЮЯЯЯ ЮДЮЙЮБЯЮБЮЏЮНЮЕЯЮБЮЙ ЮБЯЯ ЯЮЙЯ ЯЮБЯЮБЯЮЌЮНЯ ЮМЮЕЮЛЮЯЮЕЯ ЮЗ ЯЮПЯЮЎЮГЮЗЯЮЗ ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЯЮН ЮБЮНЮБЯЯЮПЮЛЮЯЮН EGFR ЯЯ ЯЯЯЯЮЗ ЮГЯЮБЮМЮМЮЎ ЯЮЕ ЯЯЮГЮКЯЮЙЯЮЗ ЮМЮЕ ЮДЮЕЯЯЮЕЯЮЗ ЮГЯЮБЮМЮМЮЎ ЮДЮЕЮН ЮЕЯЮЗЯЮЕЮЌЮЖЮЕЮЙ ЯЯЮБЯЮЙЯЯЮЙЮКЯЯ ЯЮЗЮМЮБЮНЯЮЙЮКЮЌ ЯЮЗ ЯЯ ЮНЮПЮЛЮЙЮКЮЎ ЮЕЯЮЙЮВЮЏЯЯЮЗ (OS). Ю ЯЮЯЮЕЮЙ ЯЮЕ ЮБЯ ЯЯ ЯЮП ЯЮЗЮМЮЕЮЏЮП ЮНЮБ ЯЮЗЮМЮЕЮЙЯЮИЮЕЮЏ ЯЯЮЙ ЮЮНЮБ ЮМЮЙЮКЯЯ ЯЮПЯЮПЯЯЯ ЮБЯЮИЮЕЮНЯЮН ЮМЮЕ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЯЮПЯ ЮЮЛЮБЮВЮБЮН ЮБЯЯЮЙЮКЮЌ ЯЮЗЮМЮЕЮЙЮПЮИЮЕЯЮБЯЮЕЮЏЮБ ЮДЮЕ ЮМЯЯЯЮЕЯЮБЮН ЯЮЕЮЛЮЙЮКЮЌ ЮНЮБ ЮЛЮЌЮВЮПЯ ЮН EGFR TKIs ЮЛЯЮГЯ ЯЮБЯЮЕЮЏЮБЯ ЮБЯЮЙЮДЮЕЮЏЮНЯЯЮЗЯ. ЮЯ ЯЮПЮЏ ЮПЮЙ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮЕЮНЮДЮЕЯЮПЮМЮЮНЯЯ ЮНЮБ ЮПЯЮЕЮЛЮПЯЮНЯЮБЮН ЮБЮН ЮЛЮЌЮМЮВЮБЮНЮБЮН ЯЯ ЯЯЯЯЮЗ ЮГЯЮБЮМЮМЮЎ TKI.
ЮЮЗЯЮБЮНЮЙЯЮМЮПЮЏ ЮБЮНЯЮЏЯЯЮБЯЮЗЯ ЯЯЮПЯ Я ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЯЯ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЯЮН ЯЯЮПЮН ЮКЮБЯЮКЮЏЮНЮП ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ
ЮЯЯЯ ЯЮП ЯЮЙЮП ЮКЮБЯЮБЮЛЯ ЯЮЙЮКЯ ЯЮЗЮМЮЕЮЏЮП ЯЯЯЮН ЮБЯЮПЯЮЌ ЯЮЙЯ ЯЯЮПЯЮЕЯЮПЯ ЯЮЕЯ ЮИЮЕЯЮБЯЮЕЮЏЮЕЯ ЮКЮБЮЙ ЯЮПЯ Я ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЮЕ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЮКЮБЮЙ ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЏЮНЯЮМЮБ ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ ЮЕЮЏЮНЮБЮЙ ЮЗ ЮБЮНЮЌЯЯЯ ЮОЮЗ ЮМЮЗЯЮБЮНЮЙЯЮМЯЮН ЮБЮНЯЮЏЯЯЮБЯЮЗЯ ЯЯЮПЯ Я ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЯЯ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ (ЯЯЮЎЮМЮБ 2).
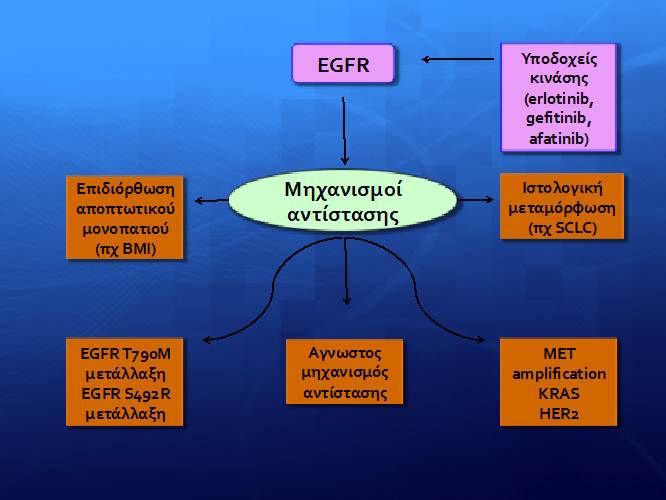
ЮЃЯЮЎЮМЮБ 2: ЮЮЗЯЮБЮНЮЙЯЮМЮПЮЏ ЮБЮНЯЮЏЯЯЮБЯЮЗЯ ЯЯЮПЯ Я ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЯЯ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ EGFR
Ю ЯЯЯЮПЯЮБЮИЮЎЯ ЮБЮНЯЮЏЯЯЮБЯЮЗ (de novo)
Ю ЮБЯЮЌ ЯЮП ЯЮЗЮМЮБЮНЯЮЙЮКЯ ЮКЮЛЮЙЮНЮЙЮКЯ ЯЯЮЕЮЛЮПЯ ЯЮПЯ ЯЯЮПЮКЯЯЯЮЕЮЙ ЮБЯЯ ЯЮЗ ЯЮПЯЮЎЮГЮЗЯЮЗ ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЯЮН ЮБЮНЮБЯЯЮПЮЛЮЯЮН ЯЮЕ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ Я ЯЮЌЯЯЮПЯ ЮН ЮЮНЮБ ЯЮПЯЮПЯЯЯ ЮБЯЮИЮЕЮНЯЮН ЯЮПЯ ЯЮЯЮПЯ ЮН ЮМЮЕЯЮЌЮЛЮЛЮБЮОЮЗ ЮБЮЛЮЛЮЌ ЮДЮЕЮН ЮПЯЮЕЮЛЮПЯЮНЯЮБЮЙ ЮБЯЯ EGFR ЯЯЮПЯЮЕЯЮПЯ ЯЮЕЯ ЮИЮЕЯЮБЯЮЕЮЏЮЕЯ ЮМЮЕ ЯЮП ЯЯЮЕЮЛЮПЯ ЯЯЮП ЮМЮЯЮП ЮДЮЙЮЌЯЯЮЗЮМЮБ PFS ЮНЮБ ЮЕЮЏЮНЮБЮЙ ЮБЯЮПЮГЮПЮЗЯЮЕЯ ЯЮЙЮКЯ ЯЮЕ ЯЯЮГЮКЯЮЙЯЮЗ ЮМЮЕ ЯЮЗ ЮКЮБЮИЮЙЮЕЯЯЮМЮЮНЮЗ ЯЮЗЮМЮЕЮЙЮПЮИЮЕЯЮБЯЮЕЮЏЮБ. ЮЅЯЮЌЯЯЮПЯ ЮН ЮДЯЮП ЮВЮБЯЮЙЮКЮПЮЏ ЮМЮЗЯЮБЮНЮЙЯЮМЮПЮЏ ЯЯЯЯЮПЯЮБЮИЮПЯЯ ЮБЮНЯЮЏЯЯЮБЯЮЗЯ: (1) ЮДЮЕЯ ЯЮЕЯЮПЯЮБЮИЮЕЮЏЯ ЮМЮЕЯЮБЮЛЮЛЮБЮГЮЯ ЯЮПЯ EGFR ЯЮПЯ ЮЕЮМЯЮПЮДЮЏЮЖЮПЯ ЮН ЯЮЗЮН ЮБЮНЮБЯЯЮПЮЛЮЎ ЯЮПЯ Я ЯЮПЮДЮПЯЮЮБ EGFR ЮБЯЯ EGFR ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЯ ЮБЮНЯЯЮПЮЛЮЮБ ЮКЮБЮЙ (2) ЮЕЯЮЙЯЯЯЯЮИЮЕЯЮЕЯ ЮГЮЕЮНЮЕЯЮЙЮКЮЯ ЮМЮЕЯЮБЮЛЮЛЮБЮГЮЯ ЯЮПЯ ЯЯ ЮНЯ ЯЮЌЯЯЮПЯ ЮН ЮМЮЕ ЯЮЗ ЮМЮЕЯЮЌЮЛЮЛЮБЮОЮЗ EGFR ЯЮЕ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЮЕ ЮМЮЗ ЮМЮЙЮКЯЮПЮКЯ ЯЯЮБЯЮЙЮКЯ ЮКЮБЯЮКЮЏЮНЮП ЯЮНЮЕЯЮМЮПЮНЮБ (ЯЮЏЮНЮБЮКЮБЯ 2).
ЮЯЯЯ ЯЯЮП 5% ЯЯЮН ЮБЯЮИЮЕЮНЯЮН ЮМЮЕ ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЏЮНЯЮМЮБ ЯЮНЮЕЯЮМЮПЮНЮБ ЮКЮБЮЙ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЮЕЮМЯЮБЮНЮЏЮЖЮЕЮЙ ЯЯЮПЯЮИЮЎЮКЮЗ (insertion) ЯЮПЯ ЮЕЮОЯЮНЮЏЮПЯ 20 ЮЗ ЮПЯЮПЮЏЮБ ЮЕЮЏЮНЮБЮЙ ЮЛЮЙЮГЯЯЮЕЯЮЗ ЮЕЯ ЮБЮЏЯЮИЮЗЯЮЗ ЯЯЮЗ ЮИЮЕЯЮБЯЮЕЮЏЮБ ЮМЮЕ EGFR ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЯЮН ЮБЮНЮБЯЯЮПЮЛЮЯЮН. ЮЯЮЙЯЯЯЯЮИЮЕЯЮБ, ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЯЮМЮЕЯЮЌЮЛЮЛЮБЮОЮЗ EGFR T790M ЯЯЮП ЮЕЮОЯЮНЮЙЮП 20 ЮЕЮМЯЮБЮНЮЏЮЖЮПЯ ЮН ЮЕЯЮЏЯЮЗЯ ЮБЮНЯЮЏЯЯЮБЯЮЗ ЯЯЮЗ ЮИЮЕЯЮБЯЮЕЮЏЮБ ЮМЮЕ EGFR TKIs. ЮЃЮЕ ЯЯЮПЮИЮЕЯЮБЯЮЕЯ ЮМЮЮНЮПЯ Я ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЮЕ T790M ЮМЮЕЯЮЌЮЛЮЛЮБЮОЮЗ, ЯЮБЮМЮЗЮЛЮЎ ЮЮКЯЯЮБЯЮЗ ЯЮПЯ BRCA1 mRNA ЯЯЮЕЯЮЏЮЖЮЕЯЮБЮЙ ЮМЮЕ ЮМЮЕЮГЮБЮЛЯЯЮЕЯЮП PFS ЮКЮБЯЮЌ ЯЮЗ ЯЮПЯЮЎЮГЮЗЯЮЗ erlotinib. ЮЄЮБ ЮДЮЕЮДЮПЮМЮЮНЮБ ЯЯЮПЯЮЕЮЏЮНЮПЯ ЮН ЯЯЮЙ ЯЮБЮМЮЗЮЛЮЌ ЮЕЯЮЏЯЮЕЮДЮБ BRCA1 ЮЕЮНЮДЮЕЯЮПЮМЮЮНЯЯ ЮНЮБ ЮБЮНЮБЮЙЯЮПЯЮН ЯЮЗ ЮБЯЮНЮЗЯЮЙЮКЮЎ ЮЕЯЮЙЯЯЮПЮЎ ЯЮЗЯ EGFR T790M ЮМЮЕЯЮЌЮЛЮЛЮБЮОЮЗЯ, ЮЕЮНЯ Я ЯЮЗЮЛЮЌ ЮЕЯЮЏЯЮЕЮДЮБ ЮЮКЯЯЮБЯЮЗЯ BRCA1 ЮНЮБ ЮПЮДЮЗЮГЮПЯЮН ЯЮЕ de novo ЮБЮНЮЌЯЯЯ ЮОЮЗ ЮБЮНЯЮЏЯЯЮБЯЮЗЯ. ЮЯЮЙЯЮЛЮЮПЮН ЯЮЗЯ EGFR T790M, ЮДЮЕЯ ЯЮЕЯЮПЯЮБЮИЮЕЮЏЯ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЮДЯЮНЮБЯЮБЮЙ ЮНЮБ ЯЯЮПЮКЮБЮЛЮЯЮПЯ ЮН ЯЯЯЯЮПЯЮБЮИЮЎ ЮБЮНЯЮЏЯЯЮБЯЮЗ (ЯЯ D761Y ЮКЮБЮЙ L858R).
Ю ЯЮБЯЮПЯ ЯЮЏЮБ ЮГЮЕЮНЮЕЯЮЙЮКЯЮН ЮМЮЕЯЮБЮЛЮЛЮБЮГЯЮН ЮЕЮЏЮНЮБЮЙ ЮДЯ ЮНЮБЯЯЮН ЮНЮБ ЮЕЮНЮПЯЮПЯЮПЮЙЮЗЮИЮЕЮЏ ЮЕЯЮЏЯЮЗЯ ЮГЮЙЮБ ЯЮЗ de novo ЮБЮНЯЮЏЯЯЮБЯЮЗ. Ю ЮЕЮНЮЕЯЮГЮПЯЮПЮЏЮЗЯЮЗ ЯЮПЯ ЮМЮПЮНЮПЯЮБЯЮЙЮПЯ PI3CA/AKT ЯЯЯЯ ЯЯЮПЮКЯЯЯЮЕЮЙ ЮМЮЕЯЮЌ ЮБЯЯ ЮБЯЯЮЛЮЕЮЙЮБ ЮЮКЯЯЮБЯЮЗЯ PTENТ ЮМЮЕЮЙЯЮНЮЕЮЙ ЯЮЗЮН ЮЕЯ ЮБЮЙЯЮИЮЗЯЮЏЮБ ЯЯЮПЯ Я EGFR TKIs. ЮЯ ЯЯЮБЯЮЙЮКЮЯ ЯЮЕЮЙЯЮЯТ ЯЮЕ ЯЯЮПЮКЮЛЮЙЮНЮЙЮКЮЯ ЮМЮЕЮЛЮЯЮЕЯТ ЮЮДЮЕЮЙЮОЮБЮН ЮЕЯЮЏЯЮЗЯ ЯЯЮЙ ЯЮП IGF1R (insulin growth factor 1 receptor) ЮМЮПЮНЮПЯЮЌЯЮЙТ ЮМЯЮПЯЮЕЮЏ ЮНЮБ ЮЕЯЮЗЯЮЕЮЌЮЖЮЕЮЙ ЯЮЗ ЮБЮНЯЮЏЯЯЮБЯЮЗ. ЮЄЮЮЛЮПЯ ЮЕЮНЮЕЯЮГЮПЯЮПЮЏЮЗЯЮЗ ЯЮПЯ NFЮКB ЯЮЎЮМЮБЯЮПЯ ЮЕЮНЮДЮЕЯЮПЮМЮЮНЯЯ ЮНЮБ ЮБЯЮПЯЮЕЮЛЮЕЮЏ ЮЮНЮБЮН ЮЕЯЮЙЯЮЛЮЮПЮН ЮМЮЗЯЮБЮНЮЙЯЮМЯ ЮБЮНЯЮЏЯЯЮБЯЮЗЯ ЯЯЮПЯ Я EGFR ЮЄЮЮЯ ЯЯЯЯ ЯЯЮПЯЮЕЮЏЮНЮПЯ ЮН ЮПЯЮЙЯЮМЮЮНЮЕЯ ЮМЮЕЮЛЮЯЮЕЯ.
|
ЮЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЮЏ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ EGFR ЯЯЮПЮН ЮКЮБЯЮКЮЏЮНЮП ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ |
|
|
Т Т Ю ЯЯЯЮПЯЮБЮИЮЎЯ ЮБЮНЯЮЏЯЯЮБЯЮЗ (de novo) |
EGFR exon 20 insertion |
BIM deletion
EGFR T790M
ЮЮНЮЕЯЮГЮПЯЮПЮЏЮЗЯЮЗ PI3CA/AKT ЮКЮБЮЙ IGF1R ЮМЮПЮНЮПЯЮБЯЮЙЮПЯ
Т
Ю ЮЏЮНЮБЮКЮБЯ 2: ЮЯЯЮЙЮПЮЙ ЮМЮЗЯЮБЮНЮЙЯЮМЮПЮЏ ЯЯЯЯЮПЯЮБЮИЮПЯЯ (de novo) ЮБЮНЯЮЏЯЯЮБЯЮЗЯ
ЮЯЮЏЮКЯЮЗЯЮЗ ЮБЮНЯЮЏЯЯЮБЯЮЗ (acquired resistance)
Ю ЯЮЙЮП ЯЯ ЯЮНЯЯ ЮЕЯЮЏЮКЯЮЗЯЮПЯ ЮМЮЗЯЮБЮНЮЙЯЮМЯЯ ЮБЮНЯЮЏЯЯЮБЯЮЗЯ ЯЯЮПЯ Я ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЯЯ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ EGFR ЯЮЕ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЮЕ ЮКЮБЯЮКЮЏЮНЮП ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ ЮКЮБЮЙ EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЮЕЮЏЮНЮБЮЙ ЮЗ ЮДЮЕЯ ЯЮЕЯЮПЮГЮЕЮНЮЎЯ ЮМЮЕЯЮЌЮЛЮЛЮБЮОЮЗ T790M, ЮЗ ЮПЯЮПЮЏЮБ ЮБЮНЮЙЯЮНЮЕЯЯЮБЮЙ ЯЯЮП 60% ЯЯЮН ЮБЯЮИЮЕЮНЯЮН ЯЮПЯ ЮЕЮМЯЮБЮНЮЏЮЖЮПЯ ЮН ЮЕЯЮЏЮКЯЮЗЯЮЗ ЮБЮНЯЮЏЯЯЮБЯЮЗ. ЮЮЙ ЮДЮЕЯ ЯЮЕЯЮПЮГЮЕЮНЮЕЮЏЯ ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЮБЯЮПЯЮЕЮЛЮПЯЮН ЮЮНЮБЮН ЯЮПЮЛЯ ЯЯ ЯЮНЯ ЮМЮЗЯЮБЮНЮЙЯЮМЯ ЮБЮНЯЮЏЯЯЮБЯЮЗЯ. Ю ЮБЯЯЮМЮПЮЙЮП ЯЮБЯЮЌЮДЮЕЮЙЮГЮМЮБ ЮБЯЮПЯЮЕЮЛЮЕЮЏ ЮЗ ABL T315I ЯЯЮЗ ЯЯЯЮНЮЙЮБ ЮМЯ ЮЕЮЛЮПЮГЮЕЮНЮЎ ЮЛЮЕЯ ЯЮБЮЙЮМЮЏЮБ (ЮЇЮЮ), ЮЗ KIT T670I ЯЯЮПЯ Я GIST (gastrointestinal stromal tumors) ЯЮГЮКЮПЯ Я ЮКЮБЮЙ ЮЗ ALK L 1196M ЯЯЮБ ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЙЮНЯЮМЮБЯЮБ. ЮЄЯЯЮП ЮЗ ЮМЮЕЯЮЌЮЛЮЛЮБЮОЮЗ T790M, ЯЯЮП ЮКЮБЮЙ ЮЗ ALK F1147L ЮЯЮПЯ ЮН ЯЮПЮН ЮЏЮДЮЙЮП ЮМЮЗЯЮБЮНЮЙЯЮМЯ ЮДЯЮЌЯЮЗЯ ЮБЯ ЮОЮЌЮНЮПЮНЯЮБЯ ЯЮЗЮН ЮБЮНЮЌЮГЮКЮЗ ЯЯЮН ЮКЮЙЮНЮБЯЯЮН ЮГЮЙЮБ ATP ЮКЮБЯЮЌ ЯЮЮНЯЮЕ ЯЮПЯЮЯ, ЮМЮЕ ЮБЯЮПЯЮЮЛЮЕЯЮМЮБ ЮНЮБ ЮМЮЕЮЙЯЮНЮЕЯЮБЮЙ ЮЗ ЮЕЯ ЮБЮЙЯЮИЮЗЯЮЏЮБ ЯЯЮН Я ЯЮПЮДЮПЯЮЯЮН erlotinib ЮКЮБЮЙ crizotinib ЮБЮНЯЮЏЯЯЮПЮЙЯЮБ.
ЮЃЮЕ ЮБЮНЯЮЏЮИЮЕЯЮЗ ЮМЮЕ ЯЮБ ЮКЯ ЯЯЮБЯЮПЯЮПЮОЮЙЮКЮЌ ЯЮЗЮМЮЕЮЙЮПЮИЮЕЯЮБЯЮЕЯ ЯЮЙЮКЮЌ ЯЮКЮЕЯ ЮЌЯЮМЮБЯЮБ ЮЗ ЮБЮНЯЮЏЯЯЮБЯЮЗ ЯЯЮЙЯ ЮБЮНЯЮЏ-EGFR ЯЯЮПЯЮЕЯЮПЯ ЯЮЕЯ ЮИЮЕЯЮБЯЮЕЮЏЮЕЯ ЯЯЮЗЮН ЮЕЮНЮЕЯЮГЮПЯЮПЮЏЮЗЯЮЗ ЮКЮБЮЙ ЮБЮЛЮЛЮЗЮЛЮЕЯЮЏЮДЯЮБЯЮЗ ЯЮПЮЛЮЛЮБЯЮЛЯЮН ЮМЮПЯЮЙЮБЮКЯЮН ЮМЮПЮНЮПЯЮБЯЮЙЯЮН (ЯЯЮЎЮМЮБ 1). ЮЄЮБ ЮМЮПЮНЮПЯЮЌЯЮЙЮБ ЮБЯ ЯЮЌ ЮБЯЮПЯЮЕЮЛЮПЯЮН ЮЮНЮБ Т ЯЮПЮЛЯЯЮЛЮПЮКЮП ЮДЮЏЮКЯЯ ЮП ЯЮПЯ ЮКЮБЮИЮПЯЮЏЮЖЮПЯ ЮН ЯЮЗЮН ЮБЮНЮЌЯЯЯ ЮОЮЗ ЯЮПЯ ЮКЯ ЯЯЮЌЯЮПЯ , ЯЮЗЮН ЮЕЯЮЙЮВЮЏЯЯЮЎ ЯЮПЯ , ЯЮЗ ЮДЮЙЮБЯЮПЯЮПЯЮПЮЏЮЗЯЮЎ ЯЮПЯ ЮКЮБЮЙ ЯЮЗЮН ЮБЯЮПЯЯЯЯЮЙЮКЮЎ ЯЮПЯ ЮЙЮКЮБЮНЯЯЮЗЯЮБ. Ю ЮЕЮНЮЕЯЮГЮПЯЮПЮЏЮЗЯЮЗ ЮКЮЌЯЮПЮЙЯЮН ЮЕЮО ЮБЯ ЯЯЮН ЯЯЮН ЮМЮПЮНЮПЯЮБЯЮЙЯЮН ЮМЯЮПЯЮПЯЮН ЮНЮБ ЮБЯЮПЯЮЕЮЛЮЯЮПЯ ЮН ЮМЮЗЯЮБЮНЮЙЯЮМЮПЯЯ ЮБЮНЯЮЏЯЯЮБЯЮЗЯ. Ю ЮЕЮНЮЏЯЯЯ ЯЮЗ ЯЮПЯ MET ЮМЮПЮНЮПЯЮБЯЮЙЮПЯ (MET amplification) ЯЮБЯЮБЯЮЗЯЮЕЮЏЯЮБЮЙ ЯЯЮП 5 ЮМЮЕ 20% ЯЯЮН ЮБЯЮИЮЕЮНЯЮН, ЮЕЮНЮЏЯЯЯ ЯЮЗ ЯЮПЯ HER2 ЮМЮПЮНЮПЯЮБЯЮЙЮПЯ (HER2 amplification) ЯЯЮП 12% ЯЯЮН ЮБЯЮИЮЕЮНЯЮН ЮКЮБЮЙ BRAF ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЯЯЮП 1% ЯЯЮН ЮБЯЮИЮЕЮНЯЮН.
Ю ЮМЮЕЯЮЌЯЯЯЯЮЗ ЯЮПЯ ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЙЮНЯЮНЮБЯЮПЯ ЯЮЕ ЮМЮЙЮКЯЮПЮКЯ ЯЯЮБЯЮЙЮКЯ ЮКЮБЮКЯЮЏЮНЯЮМЮБ ЮЗ ЮПЯЮПЮЏЮБ ЯЮБЯЮБЯЮЗЯЮЎЮИЮЗЮКЮЕ ЯЯЯЯЮЗ ЯЮПЯЮЌ ЯЮП 2010 ЮБЯЮПЯЮЕЮЛЮЕЮЏ ЮЮНЮБЮН ЮЕЯЮЙЯЯЯЯЮИЮЕЯЮП ЮМЮЗЯЮБЮНЮЙЯЮМЯ ЮБЮНЯЮЏЯЯЮБЯЮЗЯ. Ю ЮЙЯЯЮПЮЛЮПЮГЮЙЮКЮЎ ЮБЯ ЯЮЎ ЮМЮЕЯЮЌЯЯЯЯЮЗ ЮЯЮЕЮЙ ЯЮБЯЮБЯЮЗЯЮЗЮИЮЕЮЏ ЯЯЮП 3 ЮМЮЕ 15% ЯЯЮН ЯЮЕЯЮЙЯЯЯЯЮЕЯЮН ЮКЮБЮЙ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮБЯ ЯЮПЮЏ ЮМЯЮПЯЮПЯЮН ЮНЮБ ЮЕЯ ЮНЮПЮЗЮИЮПЯЮН ЮБЯЯ ЯЮЗ ЯЮПЯЮЎЮГЮЗЯЮЗ Т Т ЯЮЛЮБЯЮЏЮНЮБЯ ЮКЮБЮЙ etoposide ЮЕЯЯЯЮПЮН ЮЕЯЮЙЮВЮЕЮВЮБЮЙЯЮИЮЕЮЏ Т ЮЗ ЮБЮЛЮЛЮБЮГЮЎ ЮБЯ ЯЮЎ ЮМЮЕ ЮНЮЮБ ЮЙЯЯЮПЮЛЮПЮГЮЙЮКЮЎ ЮВЮЙЮПЯЮЏЮБ.
|
ЮЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЮЏ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ EGFR ЯЯЮПЮН ЮКЮБЯЮКЮЏЮНЮП ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ |
|
|
Т Т Т Т ЮЯЮЏЮКЯЮЗЯЮЗ ЮБЮНЯЮЏЯЯЮБЯЮЗ (Acquired resistance) |
T790M ЮМЮЕЯЮЌЮЛЮЛЮБЮОЮЗ (60%) |
MET amplification (5-20%)
HER2 amplification (12%)
BRAF ЮМЮЕЯЮЌЮЛЮЛЮБЮОЮЗ (1%)
ЮЮЕЯЮЌЯЯЯЯЮЗ ЯЮЕ SCLC (3-15%)
ЮЮГЮНЯЯЯЮПЯ ЮМЮЗЯЮБЮНЮЙЯЮМЯЯ (30%)
Т
Ю ЮЏЮНЮБЮКЮБЯ 3: ЮЯЯЮЙЮПЮЙ ЮМЮЗЯЮБЮНЮЙЯЮМЮПЮЏ ЮЕЯЮЏЮКЯЮЗЯЮЗЯ (acquired) ЮБЮНЯЮЏЯЯЮБЯЮЗЯ
ЮЃЯЯЮБЯЮЗЮГЮЙЮКЮЯ ЯЮБЯЮЌЮКЮБЮМЯЮЗЯ ЯЯЮН ЮМЮЗЯЮБЮНЮЙЯЮМЯЮН ЮБЮНЯЮЏЯЯЮБЯЮЗЯ
Т
Ю ЮБЮНЮБЮГЮНЯЯЮЙЯЮЗ ЯЯЮН ЮМЮЗЯЮБЮНЮЙЯЮМЯЮН ЮБЮНЯЮЏЯЯЮБЯЮЗЯ ЮБЯЮПЯЮЕЮЛЮЕЮЏ ЯЮП ЯЯЯЯЮП ЮВЮЎЮМЮБ ЮГЮЙЮБ ЯЮЗЮН ЮБЮНЮЌЯЯЯ ЮОЮЗ ЯЯЯЮБЯЮЗЮГЮЙЮКЯЮН ЯЮБЯЮЌЮКЮБЮМЯЮЗЯ. ЮЮЙ ЯЯЯЮБЯЮЗЮГЮЙЮКЮЯ ЮБЯ ЯЮЯ ЯЮЕЯЮЙЮЛЮБЮМЮВЮЌЮНЮПЯ ЮН ЯЯЯЮП ЮНЮЮБЯ ЮГЮЕЮНЮЙЮЌЯ EGFR ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ, ЯЯЮП ЮКЮБЮЙ ЯЯ ЮНЮДЯ ЮБЯЮМЯ ЯЯЮПЯЮЕЯ ЮПЯ ЯЯЮН ЮИЮЕЯЮБЯЮЕЮЙЯЮН.
ЮЮЮБЯ ЮГЮЕЮНЮЙЮЌЯ ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЮЏ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ
ЮЄЮП 2005, ЮПЮЙ ЮНЮЮБЯ ЮГЮЕЮНЮЙЮЌЯ ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЮЏ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ ЮДЮПЮКЮЙЮМЮЌЯЯЮЗЮКЮБЮН ЯЮЕ EGFR T790M ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ ЯЮЕ ЯЯЮП-ЮКЮЛЮЙЮНЮЙЮКЮЌ ЮМЮПЮНЯЮЮЛЮБ ЮКЯ ЯЯЮБЯЮЙЮКЯЮН ЯЮЕЮЙЯЯЮН ЮМЮЕ ЮБЮДЮЕЮНЮПЮКЮБЯЮКЮЏЮНЯЮМЮБ ЯЮНЮЕЯЮМЮПЮНЮБ. ЮЄЮП ЮЕЯЯЮМЮЕЮНЮП ЮВЮЎЮМЮБ ЮЕЯЮЙЯЮЕЯЯЮИЮЕЮЙ ЯЮП 2009 ЮМЮЕ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ ЯЮПЯ ЯЯЮПЯЮЕЯЮПЯ ЮН ЮЕЮЙЮДЮЙЮКЮЌ ЯЮЕ T790M EGFR ЮМЮЕЯЮБЮЛЮЛЮЌЮОЮЕЮЙЯ. ЮЄЮЯЮПЮЙЮПЮЙ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ ЯЮПЯ ЯЮЛЮЮПЮН ЮДЮПЮКЮЙЮМЮЌЮЖЮПЮНЯЮБЮЙ ЯЮЕ ЮКЮЛЮЙЮНЮЙЮКЮЯ ЮМЮЕЮЛЮЯЮЕЯ ЮЕЮЏЮНЮБЮЙ ЯЮПТ afatinib (BIBW 2992) ЮКЮБЮЙ ЯЮП dacomitinib (PF00299804) ЮПЮЙ ЮПЯЮПЮЏЮПЮЙ ЮКЮБЮЙ ЮПЮЙ ЮДЯЮП ЮБЮНЮБЯЯЮЮЛЮПЯ ЮН ЯЮЗ ЮДЯЮЌЯЮЗ ЮКЮБЮЙ ЯЮПЯ HER2 ЮКЮБЮЙ ЯЮПЯ HER4 ЮКЮБЮЙ ЮП CO-1686. Ю AZD9291 ЮЮДЮЕЮЙЮОЮЕ ЮЕЮЛЯЮЙЮДЮПЯЯЯЮБ ЮБЯЮПЯЮЕЮЛЮЯЮМЮБЯЮБ ЯЮЕ ЯЮЌЯЮЗЯ Ю ЮМЮЕЮЛЮЯЮЗ, ЮЕЮНЯ ЮЗ ЮБЮНЮЌЯЯЯ ЮОЮЗ ЯЯЮН neratinib (HKI-272) ЮКЮБЮЙ canertinib (CI-1033) ЮДЮЙЮБЮКЯЯЮЗЮКЮЕ ЮЮЛЮЛЮЕЮЙЯЮЗЯ ЮБЯЮПЯЮЕЮЛЮЕЯЮМЮБЯЮЙЮКЯЯЮЗЯЮБЯ ЮКЮБЮЙ ЮМЮЗ ЮБЮНЮЕЮКЯЮЎЯ ЯЮПЮОЮЙЮКЯЯЮЗЯЮБЯ. Ю ЮБЮНЮБЯЯЮПЮЛЮЮБЯ midostaurin (PKC412) ЮЮНЮБЯ ЮБЮНЮБЯЯЮПЮЛЮЮБЯ ЯЯЮН ЯЯЯЯЮЕЮЏЮНЮЗЯ ЮКЮЙЮНЮЌЯЮЗЯ C (PCK), FLT ЮКЮБЮЙ KIT ЯЮПЯ ЮГЮЙЮБ ЯЮЗЮН ЯЯЮБ ЮДЮПЮКЮЙЮМЮЌЮЖЮЕЯЮБЮЙ ЯЯЮЗЮН ЮПЮОЮЕЮЏЮБ ЮМЯ ЮЕЮПЮЛЮПЮГЮЕЮНЮЎ ЮЛЮЕЯ ЯЮБЮЙЮМЮЏЮБ ЯЮЌЮНЮЗЮКЮЕ ЯЯЮЙ ЮМЯЮПЯЮЕЮЏ ЮНЮБ ЯЯЮПЯЮЕЯЯЮЕЮЙ ЯЮЗЮН T790M ЮМЮЕЯЮЌЮЛЮЛЮБЮОЮЗ. Ю AP26113, ЮЮНЮБЯ ALK Я ЯЮПЮДЮПЯЮЮБЯ ЯЮЌЮНЮЗЮКЮЕ ЮЕЯЮЏЯЮЗЯ ЯЯЮЙ ЮДЯЮБ ЮБЮНЮБЯЯЮБЮЛЯЮЙЮКЮЌ ЮЕЯЮЏ ЯЮПЯ EGFR T790M. ЮЃЯЮЗ LUX-Lung 1 ЮКЮЛЮЙЮНЮЙЮКЮЎ ЮМЮЕЮЛЮЯЮЗ ЮБЮОЮЙЮПЮЛЮПЮГЮЎЮИЮЗЮКЮЕ ЯЮП afatinib ЯЮЕ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЯЮПЯ ЮЕЮМЯЮЌЮНЮЙЯЮБЮН ЮЕЯЮЙЮДЮЕЮЏЮНЯЯЮЗ ЮМЮЕЯЮЌ ЮБЯЯ ЯЮПЯЮЎЮГЮЗЯЮЗ erlotinib ЮЎ gefitinib. Ю ЮБЯЯЮЛЮП ЯЮПЯ ЮБЯ ЯЮЎ ЮЗ ЮМЮЕЮЛЮЯЮЗ ЮДЮЕ ЯЮБЮНЮЯЯЯЮЕ ЯЮЗЮМЮБЮНЯЮЙЮКЯ ЯЮЛЮЕЮПЮНЮЮКЯЮЗЮМЮБ ЯЯЯЮН ЮБЯЮПЯЮЌ ЯЮЗ ЯЯ ЮНЮПЮЛЮЙЮКЮЎ ЮЕЯЮЙЮВЮЏЯЯЮЗ ЯЯ ЮГЮКЯЮЙЮНЯЮМЮЕЮНЮЗ ЮМЮЕ best supportive care (HR 1.08, P=0.74), Я ЯЮЎЯЯЮЕ ЯЮЗЮМЮБЮНЯЮЙЮКЯ ЯЯЮЕЮЛЮПЯ ЯЯЮП PFS ЯЯЮП ЯЮКЮЮЛЮПЯ ЯЮПЯ afatinib (HR 0.38, P<0.0001).
ЮЃЯ ЮНЮДЯ ЮБЯЮМЯЯ ЮИЮЕЯЮБЯЮЕЮЙЯЮН
Ю ЮПЮЛЮЛЮЯ ЯЯЮЯЮПЯ ЯЮЕЯ ЮКЮЛЮЙЮНЮЙЮКЮЯ ЮМЮЕЮЛЮЯЮЕЯ ЯЮЕЯЮЙЮЛЮБЮМЮВЮЌЮНЮПЯ ЮН ЯЯ ЮНЮДЯ ЮБЯЮМЯ ЯЮПЮЛЮЛЯЮН ЯЮКЮЕЯ ЮБЯЮМЮЌЯЯЮН ЯЮПЯ ЯЯЮПЯЮЕЯЮПЯ ЮН ЯЯЮП MET ЮЎ HGF (hepatocyte growth factor), ЯЮП dasatinib, everolimus, bortezomib, bevacizumab, sunitinb ЮКЮБЮЙ cetuximab. ЮЮЯЯЮЙ ЯЯЮЙЮГЮМЮЎЯ ЯЯЯЯЯЮП ЮМЯЮНЮП ЮП ЯЯ ЮНЮДЯ ЮБЯЮМЯЯ afatinib ЮКЮБЮЙ cetuximab ЯЮБЮЏЮНЮЕЯЮБЮЙ ЮНЮБ ЯЯЮПЯЯЮЯЮЕЮЙ ЮКЮЛЮЙЮНЮЙЮКЯ ЯЯЮЕЮЛЮПЯ ЯЯЮПЮН ЮКЮБЯЮКЮЏЮНЮП ЯЮПЯ ЯЮНЮЕЯЮМЮПЮНЮБ. ЮЮНЮБЯ ЯЮЙЮИЮБЮНЯЯ ЮЛЯЮГЮПЯ ЯЮПЯ ЮДЮЕЮН ЮЯЮПЯ ЮН ЮБЮНЮЕЯ ЯЮЕЮИЮЕЮЏ ЮБЮКЯЮМЮБ ЮБЯЮПЯЮЕЮЛЮЕЯЮМЮБЯЮЙЮКЮПЮЏ ЯЯ ЮНЮДЯ ЮБЯЮМЮПЮЏ ЮЕЮЏЮНЮБЮЙ ЯЯЮЙ ЮДЮЕЮН Я ЯЮЌЯЯЮПЯ ЮН ЯЮПЮЛЮЛЮЯ ЮКЮЛЮЙЮНЮЙЮКЮЯ ЮМЮЕЮЛЮЯЮЕЯ ЯЮПЯ ЮНЮБ ЮБЯЮЕЯ ЮИЯЮНЮПЮНЯЮБЮЙ ЯЮЕ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЮЕ ЮЕЯЮЙЮВЮЕЮВЮБЮЙЯЮМЮЮНЮЗ ЮБЮНЯЮЏЯЯЮБЯЮЗ ЯЯЮПЯ Я ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЮПЯЯ ЮБЮНЮБЯЯЮПЮЛЮЕЮЏЯ. ЮЮЙт ЮБЯ ЯЯ ЮКЮБЮЙ ЮЗ ЮВЮЙЮПЯЮЏЮБ ЮМЮЕЯЮЌ ЮБЯЯ ЯЮПЯЮЎЮГЮЗЯЮЗ ЮМЮЙЮКЯЮПЮМЮПЯЮЙЮБЮКЯЮН ЮБЮНЮБЯЯЮПЮЛЮЯЮН ЯЮПЯ EGFR ЮКЮБЮЙ ЮМЮЕЯЮЌ ЮБЯЯ Я ЯЮПЯЯЮПЯЮЎ, ЮЕЮЏЮНЮБЮЙ ЮБЯЮБЯЮБЮЏЯЮЗЯЮЗ ЯЯЮПЮКЮЕЮЙЮМЮЮНЮПЯ ЮНЮБ ЮБЮНЮБЯЯЯ ЯЮИЮПЯЮН ЮБЯЮПЯЮЕЮЛЮЕЯЮМЮБЯЮЙЮКЮЯ ЮНЮЮЕЯ ЮИЮЕЯЮБЯЮЕЯ ЯЮЙЮКЮЯ ЯЯЯЮБЯЮЗЮГЮЙЮКЮЯ ЯЮЕ ЮБЯЮИЮЕЮНЮЕЮЏЯ ЮМЮЕ ЮБЮНЯЮЏЯЯЮБЯЮЗ ЯЯЮПЯ Я EGFR TKIs.
ЮЮЙЮВЮЛЮЙЮПЮГЯЮБЯЮЏЮБ
1.Т Т Т Т Т Т Т Т Howlader N, Noone AM, Krapcho M, et al. SEER Stat Fact Sheets: Lung and Bronchus. http://seercancergov/csr/1975_2008/ 2010.
2.Т Т Т Т Т Т Т Т Hopwood P, Stephens RJ. Symptoms at presentation for treatment in patients with lung cancer: implications for the evaluation of palliative treatment. The Medical Research Council (MRC) Lung Cancer Working Party. Br J Cancer 1995;71:633-6.
3.Т Т Т Т Т Т Т Т Burdett S, Stephens R, Stewart L, et al. Chemotherapy in addition to supportive care improves survival in advanced non-small-cell lung cancer: a systematic review and meta-analysis of individual patient data from 16 randomized controlled trials. J Clin Oncol 2008;26:4617-25.
4.Т Т Т Т Т Т Т Т Burdett S, Stewart L, Pignon J-P. Chemotherapy in non-small cell lung cancer: An update of an individual patient data-based meta-analysis. J Thorac Cardiovasc Surg 2005;129:1205-.
5.Т Т Т Т Т Т Т Т Rapp E, Pater JL, Willan A, et al. Chemotherapy can prolong survival in patients with advanced non-small-cell lung cancer–report of a Canadian multicenter randomized trial. J Clin Oncol 1988;6:633-41.
6.Т Т Т Т Т Т Т Т Non-small Cell Lung Cancer Collaborative Group.Т Chemotherapy in non-small cell lung cancer: a meta-analysis using updated data on individual patients from 52 randomised clinical trials. Br Med J 1995;311:899-909.
7.Т Т Т Т Т Т Т Т Fossella F, Pereira JR, von Pawel J, et al. Randomized, multinational, phase III study of docetaxel plus platinum combinations versus vinorelbine plus cisplatin for advanced non-small-cell lung cancer: the TAX 326 study group. J Clin Oncol 2003;21:3016-24.
8.Т Т Т Т Т Т Т Т Kelly K, Crowley J, Bunn PA, Jr., et al. Randomized phase III trial of paclitaxel plus carboplatin versus vinorelbine plus cisplatin in the treatment of patients with advanced non–small-cell lung cancer: a Southwest Oncology Group trial. J Clin Oncol 2001;19:3210-8.
9.Т Т Т Т Т Т Т Т Scagliotti GV, De Marinis F, Rinaldi M, et al. Phase III randomized trial comparing three platinum-based doublets in advanced non-small-cell lung cancer. J Clin Oncol 2002;20:4285-91.
10.Т Т Т Т Т Т Т Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 2002;346:92-8.
11.Т Т Т Т Т Т Т Zatloukal P, Petruzelka L, Zemanova M, et al. Gemcitabine plus cisplatin vs. gemcitabine plus carboplatin in stage IIIb and IV non-small cell lung cancer: a phase III randomized trial. Lung Cancer 2003;41:321-31.
12.Т Т Т Т Т Т Т Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129-39.
13.Т Т Т Т Т Т Т Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497-500.
14.Т Т Т Т Т Т Т Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693-703.
15.Т Т Т Т Т Т Т Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 2012;30:863-70.
16.Т Т Т Т Т Т Т Ou SI, Bang Y, Camidge DR, et al. Efficacy and safety of crizotinib in patients with advanced ROS1-rearranged non-small cell lung cancer (NSCLC). J Clin Oncol 2013;Suppl:abstr 8032.
17.Т Т Т Т Т Т Т Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008;359:1367-80.
18.Т Т Т Т Т Т Т Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007;7:169-81.
19.Т Т Т Т Т Т Т Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947-57.
20.Т Т Т Т Т Т Т Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239-46.
21.Т Т Т Т Т Т Т Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735-42.
22.Т Т Т Т Т Т Т Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010;362:2380-8.
23.Т Т Т Т Т Т Т Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121-8.
24.Т Т Т Т Т Т Т Yang JC-H, Schuler MH, Yamamoto N, et al. LUX-Lung 3: A randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations. J Clin Oncol 2012;30:LBA7500.
25.Т Т Т Т Т Т Т Wu YL, Zhou C, Hu C, et al. LUX-Lung 6: A randomized, open-label, phase III study of afatinab (A) versus gemcitabine/cisplatin (GC) as first-line treatment for Asian patients (pts) with EGFR mutation-positive (EGFR M+) advanced adenocarcinoma of the lung. J Clin Oncol 2013;31:abstr 8016.
26.Т Т Т Т Т Т Т Fukuoka M, Wu YL, Thongprasert S, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol 2011;29:2866-74.
27.Т Т Т Т Т Т Т Mok T, Yang JJ, Lam KC. Treating patients with EGFR-sensitizing mutations: first line or second line–is there a difference? J Clin Oncol 2013;31:1081-8.
28.Т Т Т Т Т Т Т J. Y. Wu, S. G. Wu, C. H. Yang et al., тLung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response,т Clinical Cancer Research, vol. 14, no. 15, pp. 4877т4882, 2008.
29.Т Т Т Т Т Т Т S. Maheswaran, L. V. Sequist, S. Nagrath et al., тDetection of mutations in EGFR in circulating lung-cancer cells,т New England Journal of Medicine, vol. 359, no. 4, pp. 366т377, 2008.
30.Т Т Т Т Т Т Т M. Inukai, S. Toyooka, S. Ito et al., тPresence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer,т Cancer Research, vol. 66, no. 16, pp. 7854т7858, 2006.
31.Т Т Т Т Т Т Т L. Prudkin, X. Tang, and I. I. Wistuba, тGerm-line and somatic presentations of the EGFR T790M mutation in lung cancer,т Journal of Thoracic Oncology, vol. 4, no. 1, pp. 139т141, 2009.
32.Т Т Т Т Т Т Т R. Rosell, M. A. Molina, C. Costa et al., тPretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations,т Clinical Cancer Research, vol. 17, no. 5, pp. 1160т1168, 2011.
33.Т Т Т Т Т Т Т M. L. Sos, M. Koker, B. A. Weir et al., тPTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of akt and EGFR,т Cancer Research, vol. 69, no. 8, pp. 3256т3261, 2009.
34.Т Т Т Т Т Т Т I. Vivanco, D. Rohle, M. Versele et al., тThe phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation,т Proceedings of the National Academy of Sciences of the United States of America, vol. 107, no. 14, pp. 6459т6464, 2010.
35.Т Т Т Т Т Т Т Y. Ben-Neriah and M. Karin, тInflammation meets cancer, with NF-ЮКB as the matchmaker,т Nature Immunology, vol. 12, no. 8, pp. 715т723, 2011.
36.Т Т Т Т Т Т Т T. G. Bivona, H. Hieronymus, J. Parker et al., тFAS and NF-ЮКB signalling modulate dependence of lung cancers on mutant EGFR,т Nature, vol. 471, no. 7339, pp. 523т526, 2011.
37.Т Т Т Т Т Т Т Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res 2011;17:1169-80.
38.Т Т Т Т Т Т Т Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19:2240-7.
39.Т Т Т Т Т Т Т Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov 2012;2:922-33.
40.Т Т Т Т Т Т Т Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A 2012;109:E2127-33.
41.Т Т Т Т Т Т Т Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26.
42.Т Т Т Т Т Т Т Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007;104:20932-7.
43.Т Т Т Т Т Т Т Engelman JA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039-43.
44.Т Т Т Т Т Т Т Bridges, A.J. The rationale and strategy used to develop a series of highly potent, irreversible, inhibitors of the epidermal growth factor receptor family of tyrosine kinases. Curr. Med. Chem. 6, 825т843 (1999).
45.Т Т Т Т Т Т Т Kwak, E. The role of irreversible HER family inhibition in the treatment of patients with non-small cell lung cancer. Oncologist 16, 1498т1507 (2011).
46.Т Т Т Т Т Т Т Kobayashi, S. et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786т792 (2005).
47.Т Т Т Т Т Т Т Kwak, E.L. et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 102, 7665т7670 (2005).
48.Т Т Т Т Т Т Т Zhou, W. et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070т1074 (2009).
49.Т Т Т Т Т Т Т Dienstmann, R., De Dosso, S., Felip, E. & Tabernero, J. Drug development to overcome resistance to EGFR inhibitors in lung and colorectal cancer. Mol. Oncol. 6, 15т26 (2012).
50.Т Т Т Т Т Т Т Yu, H.A. & Riely, G.J. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in lung cancers. J. Natl. Compr. Canc. Netw. 11, 161т169 (2013).
51.Т Т Т Т Т Т Т Ranson, M. et al. Preliminary results from a Phase I study with AZD9291: an irreversible inhibitor of epidermal growth factor receptor (EGFR) activating and resistance mutations in non-small cell lung cancer (NSCLC). European Cancer Conference (Amsterdam, 2013).
52.Т Т Т Т Т Т Т Ou, S.H. Second-generation irreversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs): a better mousetrap? A review of the clinical evidence. Crit. Rev. Oncol. Hematol. 83, 407т421 (2012).
53.Т Т Т Т Т Т Т Lee, H.J. et al. Noncovalent wild-type-sparing inhibitors of EGFR T790M. Cancer Discov. 3, 168т181 (2013).
54.Т Т Т Т Т Т Т Rivera, V.M. et al. AP26113 is a dual ALK/EGFR inhibitor: characterization against EGFR T790M in cell and mouse models of NSCLC. Cancer Res. 72 (suppl. 1), abstract 1794 (2012).
55.Т Т Т Т Т Т Т Janjigian, Y. et al. Activity and tolerability of afatinib (BIBW 2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. J. Clin. Oncol. 29, 7525 (2011).
56.Т Т Т Т Т Т Т Janjigian, Y.Y. et al. Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin. Cancer Res. 17, 2521т2527 (2011).
57.Т Т Т Т Т Т Т Robinson, K.W. & Sandler, A.B. The role of MET receptor tyrosine kinase in non-small cell lung cancer and clinical development of targeted anti-MET agents. Oncologist 18, 115т122 (2013).
58.Т Т Т Т Т Т Т Johnson, M.L. et al. Phase II trial of dasatinib for patients with acquired resistance to treatment with the epidermal growth factor receptor tyrosine kinase inhibitors erlotinib or gefitinib. J. Thorac. Oncol. 6, 1128т1131 (2011).
59.Т Т Т Т Т Т Т Riely, G.J. et al. Prospective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimus. Clin. Cancer Res. 13, 5150т5155 (2007).
60.Т Т Т Т Т Т Т Lynch, T.J. et al. A randomized phase 2 study of erlotinib alone and in combination with bortezomib in previously treated advanced non-small cell lung cancer. J. Thorac. Oncol. 4, 1002т1009 (2009).
61.Т Т Т Т Т Т Т Herbst, R.S. et al. Efficacy of bevacizumab plus erlotinib versus erlotinib alone in advanced non-small-cell lung cancer after failure of standard first-line chemotherapy (BeTa): a double-blind, placebo-controlled, phase 3 trial. Lancet 377, 1846т1854 (2011).
62.Т Т Т Т Т Т Т Scagliotti, G.V. et al. Sunitinib plus erlotinib versus placebo plus erlotinib in patients with previously treated advanced non-small-cell lung cancer: a phase III trial. J. Clin. Oncol. 30, 2070т2078 (2012).






