 Γενετική και Καρκίνος παχέος εντέρου
Γενετική και Καρκίνος παχέος εντέρου
Ιωάννης Σουγκλάκος
Επίκουρος Καθηγητής Παθολογίας-Ογκολογίας
Πανεπιστήμιο Κρήτης
ΕΙΣΑΓΩΓΗ
Ο κολοορθικός καρκίνος (colorectal cancer – CRC) είναι μία συνήθης και θανατηφόρος νόσος. Περίπου 145.290 νέες περιπτώσεις διαγιγνώσκονται κάθε χρόνο στις Ηνωμένες Πολιτείες, από τις οποίες οι 104.950 προέρχονται από το κόλον και οι υπόλοιπες από το ορθό. Το 2005, περίπου 56.290 Αμερικανοί αναμένεται να πεθάνουν από κολοορθικό καρκίνο, αντιστοιχώντας περίπου στο 10% των θανάτων λόγω νεοπλασίας. Ο κολοορθικός καρκίνος αποτελεί την τρίτη αιτία θανάτου από καρκίνο τόσο στους άνδρες, όσο και στις γυναίκες.
Οι παράγοντες κινδύνου για τον κολοορθικό καρκίνο είναι τόσο περιβαλλοντικοί, όσο και γενετικοί. Ο τρόπος εμφανίσεώς του ακολουθεί συνήθως ένα από τα ακόλουθα τρία πρότυπα, του σποραδικού, του κληρονομουμένου και του οικογενούς, αναλόγως των παραγόντων κινδύνου οι οποίοι ενοχοποιούνται για κάθε μορφή.
Σποραδική Μορφή
Η σποραδική μορφή, αποτελεί περίπου στο 70% όλων των περιπτώσεων και δεν υπάρχει οικογενειακό ιστορικό. Κατά κανόνα αφορά ασθενείς ηλικίας μεγαλύτερης των 50 ετών. Διατροφικοί και περιβαλλοντικοί παράγοντες έχουν συσχετισθεί με την εμφάνιση της νόσου.
Κληρονομική Μορφή
Η κληρονομική μορφή αφορά σε περίπου 5% των ασθενών και αυτές οι περιπτώσεις υποδιαιρούνται αναλόγως με τον αν η ανεύρεση πολυπόδων αποτελεί ή όχι την κυρία εκδήλωση της νόσου. Οι ασθένειες με πολυποδίαση περιλαμβάνουν τα οικογενή σύνδρομα αδενωματώδους πολυποδιάσεως (οικογενής αδενωματώδης πολυποδίαση, FAP: Familial Adenomatous Polyposis, το σύνδρομο Gardner και το σύνδρομο Turcot) καθώς και τα οικογενή σύνδρομα αμαρτωματώδους πολυποδιάσεως (Peutz-Jeghers, νεανική πολυποδίαση, νόσος Cowden)2. Η εμφάνιση κληρονομουμένου κολοορθικού καρκίνου χωρίς την ταυτόχρονη ανεύρεση πολλαπλών πολυπόδων αποτελεί διακριτή νοσολογική οντότητα, γνωστή ως κληρονομικός μη-πολυποσικός κολοορθικός καρκίνος (Hereditary Non-Polyposis Colon Cancer, HNPCC ή σύνδρομο Lynch). Το σύνδρομο συνοδεύεται από υψηλό κίνδυνο αναπτύξεως καρκίνου του παχέος εντέρου, ενώ οι γενετικές μεταλλαγές οι οποίες το χαρακτηρίζουν έχουν αναγνωριστεί.
Τα οικογενή πολυποσικά σύνδρομα είναι περιλαμβάνουν τα «οικογενή σύνδρομα αδενωματώδους πολυποδιάσεως» και τα «οικογενή σύνδρομα αμαρτωματώδους πολυποδιάσεως».
Οικογενή Σύνδρομα Αδενωματώδους Πολυποδιάσεως
Α. Το «σύνδρομο οικογενούς αδενωματώδους πολυποδιάσεως-ΣΟΑΠ» χαρακτηρίζεται από την παρουσία εκατοντάδων έως χιλιάδων πολυπόδων σε όλη την έκταση του παχέος εντέρου με ιστολογικά χαρακτηριστικά τυπικών αδενωμάτων. Η μέση ηλικία αναπτύξεως πολυπόδων είναι τα 25 έτη. Η μέση ηλικία εμφανίσεως συμπτωμάτων είναι 33 έτη, ενώ η μέση ηλικία διαγνώσεως των πολυπόδων τα 36 έτη. Η μέση ηλικία διαγνώσεως διηθητικού καρκίνου είναι τα 42 έτη. Το ΣΟΑΠ συνοδεύεται επίσης από μια πληθώρα εξωεντερικών εκδηλώσεων, οι οποίες περιλαμβάνουν οστεώματα της γνάθου, πολύποδες του ανωτέρου πεπτικού σωλήνα, συγγενή υπερτροφία του επιθηλίου του αμφιβληστροειδούς, καθώς και αυξημένο κίνδυνο εμφανίσεως άλλων νεοπλασμάτων, όπως ο καρκίνος του φύματος του Vater, ο θηλώδης και θηλακιώδης καρκίνος του θυρεοειδούς, ο γαστρικός καρκίνος, και το μυελοβλάστωμα και ηπατοβλάστωμα στα παιδιά. Η γενετική διαταραχή, οποία ευθύνεται για το σύνδρομο, είναι η γαμετική μετάλλαξη του γονιδίου APC, η οποία κληρονομείται κατά τον αυτόσωμο επικρατούντα τύπο με υψηλή διεισδυτικότητα. Συγκεκριμένες μεταλλάξεις σχετίζονται με την προεξάρχουσα κλινική εικόνα. Επί παραδείγματι, μεταλλάξεις στα κωδώνια 1250-1464 συνοδεύονται από εμφάνιση χιλιάδων πολυπόδων, ενώ μεταλλάξεις στο κωδώνιο 1465-1466 προδιαθέτουν στην εμφάνιση εξωεντερικών εκδηλώσεων. Ασθενείς με θετικό ιστορικό για ΣΟΑΠ πρέπει να ελέγχονται για μετάλλαξη του APC και, επί θετικού αποτελέσματος, να υποβάλλονται σε έλεγχο με ενδοσκόπηση από την ηλικία των 12 ετών. Η μόνη ριζική θεραπευτική επιλογή είναι η ολική κολεκτομή και είναι προτιμότερο οι ασθενείς αυτοί να αντιμετωπίζονται σε κέντρα εξειδικευμένα στην γενετική καθοδήγηση.
Β. Το «εξασθενημένο ΣΟΑΠ» αποτελεί μια παραλλαγή τού κλασσικού ΣΟΑΠ, στην οποία οι ασθενείς εμφανίζουν μικρότερο αριθμό πολυπόδων (εξ ορισμού <100) και αναπτύσσουν καρκίνου σε μεγαλύτερη ηλικία (περίπου 12 έτη αργότερα) σε σχέση με τους ασθενείς με κλασσικό ΣΟΑΠ. Το εξασθενημένο ΣΟΑΠ οφείλεται σε μετάλλαξη του APC σε περιοχές κοντά στο 5’ (κωδώνιο 158) και 3’ (κωδώνιο 1596). Ο μέσος αριθμός πολυπόδων είναι 25, η διάμεση ηλικία διαγνώσεως καρκίνου είναι 58 έτη, ενώ το 69% των ασθενών εμφανίζει κολοορθικό καρκίνο έως την ηλικία των 80 ετών.
Γ. Το «σύνδρομο Gardner» περιλαμβάνει τις ίδιες εκδηλώσεις από το έντερο, όπως το ΣΟΑΠ, οι εξωεντερικές όμως εκδηλώσεις είναι περισσότερο έκδηλες και ποικίλες και περιλαμβάνουν επιπροσθέτως οστεώματα των μακρών οστών, δεσμοειδείς όγκους και ίνωση του μεσεντερίου, η οποία καταλήγει σε θάνατο στο 8-11% των περιπτώσεων.
Δ. Το «σύνδρομο Turcot» χαρακτηρίζεται από την ανάπτυξη νεοπλασμάτων στο παχύ έντερο και το κεντρικό νευρικό σύστημα. Δύο διαφορετικοί τύποι του συνδρόμου έχουν περιγραφεί: α) ΣΟΑΠ και πολύμορφο γλοιοβάστωμα λόγω μεταλλάξεων του APC και β) καρκίνος παχέος εντέρου και αστροκύττωμα λόγω μεταλλάξεων των γονιδίων διορθώσεως αποτυχημένου συνταιριάσματος (MMR: miss match repair)
Οικογενή Σύνδρομα Αμαρτωματώδους Πολυποδιάσεως
Α. Το «σύνδρομο Peutz-Jeghers» χαρακτηρίζεται από την εναπόθεση μελανίνης πέριξ των χειλέων, την μύτη, τον στοματικό βλεννογόνο, τα πέλματα και τις παλάμες. Χαρακτηρίζεται από την παρουσία αμαρτωμάτων στο πεπτικό σύστημα (στόμαχος, λεπτό έντερο, κόλον), τα οποία δυνατό να εκδηλωθούν με αιμορραγία, απόφραξη ή εγκολεασμό. Περίπου το 50% των ασθενών αναπτύσσει ορθοκολικό ή γαστρικό καρκίνο, νεοπλάσματα λεπτού εντέρου, και σπανίως παγκρεατικό καρκίνος, με μέση ηλικία διαγνώσεως τα 50 έτη. Ωοθικές κύστεις και νεοπλάσματα εμβρυϊκής προελεύσεως των ωοθηκών αναφέρονται στο 5-12% των περιπτώσεων. Ως υπεύθυνη γενετική μεταβολή έχει προσφάτως χαρακτηριστεί η γαμετική μετάλλαξη ενός γονιδίου, το οποίο εδράζεται στο χρωμόσωμα 19p13.3 και κωδικοποιεί μια κινάση με δράση σερίνης/θρεονίνης (LKB1 ή STK11). Η ακριβής δράση αυτής της κινάσης δεν είναι ακόμη γνωστή και υποστηρίζεται ότι πιθανώς να είναι ογκοκατασταλτική. Ο γενετικός έλεγχος είναι πλέον εφικτός σε εξειδικευμένα εργαστήρια.
Β. Το «σύνδρομο νεανικής πολυποδιάσεως» χαρακτηρίζεται από την ανεύρεση αμαρτωμάτων κυρίως του ορθού, σε παιδική ηλικία, τα οποία εκδηλώνονται με αιμορραγία. Προσφάτως, έχουν περιγραφεί τρεις μορφές του συνδρόμου: α) η οικογενής νεανική πολυποδίαση του κόλου, β) η οικογενής νεανική πολυποδίαση του στομάχου και γ) η γενικευμένη νεανική πολυποδίαση. Γαμετικές μεταλλάξεις στο SMAD4 (ένα μέλος της οικογενείας του υποδοχέα του TGF-beta) έχουν εντοπιστεί, σε ασθενείς με νεανική πολυποδίαση.
Γ. Η «νόσος του Cowden» (σύνδρομο πολλαπλών αμαρτωμάτων) χαρακτηρίζεται από πολλαπλούς αμαρτωματώδεις όγκους εκτοδερμικής, μεσοδερμικής και ενδοδερμικής προελέυσεως. Η νόσος συσχετίζεται με αυξημένο κίνδυνο αναπτύξεως καρκίνου του μαστού, θυρεοειδοπάθειες, υποδόρια λιπώματα, λειομυώματα της μήτρας και πολύποδες του πεπτικού συστήματος. Ως υπέυθυνη γονιδιακή βλάβη έχει χαρακτηριστεί η μετάλλαξη του PTEN γονιδίου, η οποία οδηγεί σε συνεχή ενεργοποίηση της οδού μεταδόσεως σήματος μέσω του PI3K/Akt.
Οικογενής Μορφή
Το λιγότερο κατανοητό πρότυπο αναπτύξεως κολοορθικού καρκίνου είναι το οικογενές. Έως το 25% των περιπτώσεων υπάγεται σε αυτή την κατηγορία. Οι προσβεβλημένοι ασθενείς έχουν οικογενειακό ιστορικό κολοορθικού καρκίνου, αλλά η μορφή δεν εντάσσεται σε κάποιο από τα κληρονομούμενα σύνδρομα, τα οποία έχουν ήδη αναφερθεί. Άτομα από τις συγκεκριμένες οικογένειες παρουσιάζουν υψηλό κίνδυνο αναπτύξεως καρκίνου του παχέος εντέρου, ο οποίος όμως είναι σημαντικός χαμηλότερος από τον αντίστοιχο των κληρονομουμένων συνδρόμων. Η ύπαρξη ενός πρώτου βαθμού προσβεβλημένου συγγενή (π.χ. γονέα, παιδί ή αδελφό) συνεπάγεται αύξηση κινδύνου αναπτύξεως κολοορθικού καρκίνου κατά 1.7 φορές, σε σύγκριση με τον γενικό πληθυσμό. Ο κίνδυνος αυξάνεται επιπλέον εάν δύο συγγενείς πρώτου βαθμού έχουν καρκίνο του παχέος εντέρου ή εάν το πρώτο περιστατικό στην οικογένεια έχει διαγνωστεί πριν από την ηλικία των 55 ετών.
Ο βαθμός κατανοήσεως των μοριακών γεγονότων, τα οποία χαρακτηρίζουν τον καρκίνο του παχέος εντέρου είναι κατά πολύ μεγαλύτερος από εκείνον των άλλων συμπαγών όγκους. Συγκεκριμένες μεταλλαγές βασικού γενετικού υλικού είναι υπεύθυνες για το κληρονομούμενο σύνδρομο κολοορθικού καρκίνου ενώ μία σταδιακή συσσώρευση σωματικών μεταλλαγών χαρακτηρίζει τις περισσότερες σποραδικές περιπτώσεις. Αντιθέτως, οι γενετικές ανωμαλίες οι οποίες χαρακτηρίζουν την οικογενή νόσο παραμένουν ατελώς κατανοητές. Τουλάχιστον σε ασθενείς καταγομένους από τους Εβραίους Ashkenazi, μία συγκεκριμένη μεταλλαγή στο γονίδιο του αδενωματώδους πολύποδα (APC: adenomatous polyposis coli) συνδέεται με ένα οικογενή τύπο κολοορθικού καρκίνου. Άλλοι ερευνητές έχουν δείξει ότι αυτές οι οικογένειες αντιπροσωπεύουν μία παραλλαγή του HNPCC3 και συνεπώς ανωμαλίες των διορθωτικών γονιδίων συνταιριάσματος του DNA εμπλέκονται σε σημαντικό ποσοστό αυτών των ασθενειών4-6.
Η ανίχνευση συγκεκριμένων γενετικών μεταλλαγών υπευθύνων για την ογκογένεση στο παχύ έντερο έχει άμεση επίδραση στην κλινική μέριμνα. Οι ασθενείς με υψηλότερο κίνδυνο αναπτύξεως κολοορθικού καρκίνου ανιχνεύονται μέσω γενετικού ελέγχου για ειδικές μεταλλαγές βασικού γενετικού υλικού, ενώ νέες μοριακές μέθοδοι ελέγχου πρόωρης ανευρέσεως καρκίνου του παχέος εντέρου με ανίχνευση μεταλλαγών σε υλικό κοπράνων είναι υπό μελέτη. Επιπλέον, αυτές οι μεταλλαγές ελέγχονται επίσης ως πιθανοί θεραπευτικοί στόχοι.
ΓΟΝΙΔΙΑΚΕΣ ΜΕΤΑΛΛΑΞΕΙΣ ΚΑΙ ΚΑΡΚΙΝΟΓΕΝΕΣΗ ΠΑΧΕΟΣ ΕΝΤΕΡΟΥ
Σε αυτό το κεφάλαιο επιχειρείται μία επισκόπηση των κυριοτέρων γενετικών αλλαγών, οι οποίες θεωρούνται υπεύθυνες για την καρκινογένεση του παχέος εντέρου, τόσο σε ασθενείς με σποραδικό καρκίνο όσο και σε ασθενείς με κληρονομούμενα σύνδρομα πολυποδιάσεως. Οι κληρονομικοί παράγοντες οι οποίοι σχετίζονται με τον HNPCC και ο ρόλος στρατηγικών γενετικών ελέγχων και εξετάσεων για τους ασθενείς αυτούς συζητούνται αναλυτικώς σε επόμενη ενότητα.
Οι γενετικές μεταλλάξεις είναι κληρονομικές ή επίκτητες. Κάθε γενετική μετάλλαξη η οποία συμβαίνει πριν ή κατά την γονιμοποίηση του ωαρίου καλείται μετάλλαξη του βασικού γενετικού υλικού και μεταδίδεται από τον γονέα στον απόγονο ως κληρονομικό ελάττωμα. Αν η μετάλλαξη συμβεί αιφνιδίως στο σπέρμα, το ωάριο ή τον ζυγώτη, οι γονείς του προσβληθέντος προσώπου δεν παρουσιάζουν το φαινότυπο του καρκίνου, αλλά οι μελλοντικοί απόγονοι είναι δυνατό να κληρονομήσουν την νέα-de novo μετάλλαξη. Συχνότερα, μία μετάλλαξη εμφανίζεται σε ένα κύτταρο κατά την αύξηση ή την ανάπτυξη ενός συγκεκριμένου ιστού ή οργάνου. Η μετάλλαξη αυτή καλείται σωματική. Επειδή αυτές οι μεταλλάξεις συχνά επιφέρουν ένα επιλεκτικό πλεονέκτημα αναπτύξεως, έχουν ως αποτέλεσμα τον πολλαπλασιασμό του κυττάρου το οποίο περιέχει το μεταλλαγμένο γενετικό υλικό (κλωνική εξέλιξη του καρκίνου)7.
Η κλωνική φύση των όγκων είναι ένα κρίσιμο χαρακτηριστικό της θεωρίας της σωματικής μεταλλάξεως / κλωνικής εξελίξεως της ανθρώπινης καρκινογενέσεως. Σύμφωνα με αυτό το πρότυπο, το πλεονέκτημα αναπτύξεως το οποίο αποκτάται από ένα μοναδικό μεταλλαγμένο κύτταρο επιτρέπει στους απογόνους του να υπερισχύουν αριθμητικώς των γειτονικών μη μεταλλαγμένων κυττάρων. Μέσα από αυτόν τον πληθυσμό, ένα μοναδικό κύτταρο υφίσταται μία δεύτερη μετάλλαξη, εξασφαλίζοντας ένα επιπλέον πλεονέκτημα αναπτύξεως το οποίο επιτρέπει επιπλέον κλωνική επέκταση. Διαδοχικά κύματα κλωνικής επεκτάσεως χαρακτηρίζονται από την διαδοχική απόκτηση επιπλέον μεταλλάξεων, την επιπλέον κυτταρική αποδιοργάνωση και τελικώς την ικανότητα διηθήσεως και μεταστάσεως.
Η Διαδοχή Αδενώματος – Καρκινώματος
Οι περισσότεροι καρκίνοι του παχέος εντέρου του ανθρώπου πιστεύεται ότι εκδηλώνονται από αδενώματα του εντέρου (αδενωματώδεις πολύποδες), τα οποία είναι δυσπλαστικά αλλά όχι κακοήθη (εικόνα 1). Οι αδενωματώδεις πολύποδες σχηματίζονται στο κόλον, όταν οι φυσιολογικοί μηχανισμοί οι οποίο ελέγχουν την επιθηλιακή ανανέωση απορυθμιστούν. Τα επιφανειακά κύτταρα κατά μήκος του εντέρου συνεχώς αποπίπτουν στον εντερικό σωλήνα και πρέπει συνεχώς να αντικαθίστανται. Ο πολλαπλασιασμός λαμβάνει χώρα αποκλειστικώς στις κρύπτες. Καθώς τα κύτταρα προωθούνται στην επιφάνεια του αυλού, παύουν να πολλαπλασιάζονται και τελικώς διαφοροποιούνται. Αυτή η διαδικασία σταδιακώς απορυθμίζεται, καθώς τα αδενώματα αυξάνουν σε μέγεθος, καθίστανται δυσπλαστικά και τελικώς αποκτούν διηθητικό δυναμικό.
Η υπόθεση ότι οι διηθητικό νεόπλασμα αναπτύσσεται από ενδιάμεσες, προκαρκινικές, πρόδρομες μορφές στηρίζεται σε παθολογοανατομικά, επιδημιολογικά και κλινικά δεδομένα, τόσο σε ανθρώπινα όσο και σε πειραματικά πτότυπα πειραματοζώων. Συνοπτικώς:
- πρώιμοι καρκίνοι παρατηρούνται συχνά σε μεγάλους αδενωματώδεις πολύποδες, ενώ περιοχές αδενωματώδους αλλαγής συχνά ανευρίσκονται πέριξ των διηθητικών νεοπλασμάτων,
- τα αδενώματα και τα καρκινώματα παρουσιάζουν παρόμοια κατανομή σε όλη την έκταση του εντέρου και αδενώματα τυπικώς ανευρίσκονται 10-15 χρόνια πριν την εμφάνιση καρκίνου, τόσο της σποραδικής όσο και της οικογενούς μορφής,
- σε πρότυπα πειραματοζώων με καρκίνο παχέος εντέρου, αδενώματα αναπτύσσονται πριν την ανάπτυξη καρκινωμάτων, τα οποία αναπτύσσονται ομοιομόρφως στον αδενωματώδη ιστό,
- η σημαντική μείωση της συχνότητας εμφανίσεως καρκίνου του παχέος εντέρου μετά την αφαίρεση των πολυπόδων έχει αποδειχθεί σε τυχαιοποιημένες κλινικές μελέτες σε ανθρώπους8.
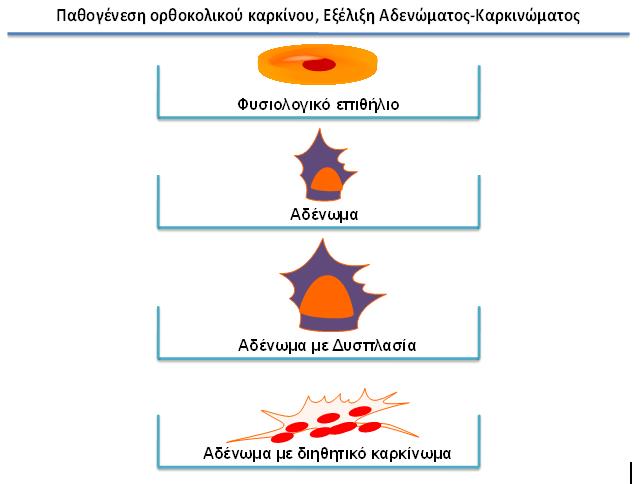
Εικόνα 1: Παθογενετικά στάδια του κολοορθικού καρκίνου.
Η Διαδικασία Πολλαπλών Βημάτων της Καρκινογενέσεως του Παχέος Εντέρου
Πιστεύεται ότι συγκεκριμένες γενετικές αλλαγές οδηγούν στον μετασχηματισμό του φυσιολογικού επιθηλίου του παχέος εντέρου σε διηθητικό καρκίνο. Το 1990, οι Fearon και Vogelstein7 περιέγραψαν την μοριακή βάση αναπτύξεως του καρκίνου του παχέος εντέρου ως μία διαδικασία πολλαπλών βημάτων, στα οποία κάθε συσσωρευόμενο γενετικό συμβάν προσφέρει ένα επιλεκτικό πλεονέκτημα αναπτύξεως στο επιθηλιακό κύτταρο του παχέος εντέρου. Μεταγενέστερες μελέτες να επιβεβαιώσουν και να βελτιώσουν την ανωτέρω υπόθεση.
Σύμφωνα με το μοντέλο Fearon και Vogelstein, απαιτούνται μεταλλάξεις του βασικού γενετικού υλικού ή σωματικές μεταλλάξεις για τον κακοήθη μετασχηματισμό, και είναι η συσσώρευση πολλαπλών γενετικών μεταλλάξεων παρά η ακολουθία τους, η οποία τελικώς καθορίζει την βιολογική συμπεριφορά του όγκου (εικόνα 2). Οι μεταλλάξεις του βασικού γενετικού υλικού χαρακτηρίζουν τα κοινά κληρονομικά σύνδρομα (ΣΟΑΠ, HNPCC), ενώ οι σποραδικοί καρκίνοι προέρχονται από την σταδιακή συσσώρευση πολλαπλών σωματικών μεταλλάξεων. Οι μεταλλάξεις στο γονίδιο APC, οι οποίες είναι κοινό χαρακτηριστικό και στους κληρονομικούς και στους σποραδικούς όγκους, εμφανίζονται νωρίς στην διαδικασία ογκογενέσεως, ενώ η μεταλλάξεις του κατασταλτικού γονιδίου p53 λαμβάνουν χώρα σε απώτερα στάδια της διαδικασίας.
Οι συγκεκριμένες μεταλλάξεις οι οποίες εμπλέκονται στην ανθρώπινη ογκογένεση περιλαμβάνουν σημειακές μεταλλάξεις, αλλαγή της μεθυλιώσεως του DNA και γονιδιακές αναδιατάξεις, διπλασιασμούς και διαγραφές. Αυτές οι μεταλλάξεις ομαδοποιούνται αναλόγως γενετικών μοριακών τους συνεπειών, ως ακολούθως:
- μεταλλάξεις αποκτήσεως λειτουργίας εμπλέκουν την ενεργοποίηση οδών οι οποίες προωθούν την κακοήθη εξαλλαγή του κυττάρου, συμπεριλαμβανομένης και της ενεργοποιήσεως των ογκογονιδίων
- μεταλλάξεις απωλείας λειτουργίας, οι οποίες συνήθως εμπλέκουν ογκοκατασταλτικά γονίδια ή αποπτωτικές οδούς
- επιγενείς μεταβολές, όπως η μεθυλίωση του DNA και η απώλεια μεταγραφής, η οποία δυνατό οδηγήσει στην μη έκφραση συγκεκριμένων γονιδίων μέσω της μεθυλιώσεως των εκκινητών των9.
Υπάρχουν ενδείξεις ότι και οι τρεις ως άνω μηχανισμοί εμπλέκονται στην παθογένεση του καρκίνου του παχέος εντέρου. Η μεθυλίωση της περιοχής του εκκινητή μερικών γονιδίων επιδιορθώσεως βλαβών του DNA (MMR: missmatch repair) με αποτέλεσμα την απώλεια της εκφράσεως αυτών των γονιδίων θεωρείται ότι αποτελεί την βάση αναπτύξεως του σποραδικού κολοορθικού καρκίνου και μερικών περιπτώσεων HNPCC.
ΠΙΝΑΚΑΣ 1
Μεταλλάξεις Γονιδίων, Εμπλεκομένων στην Ανάπτυξη του Καρκίνου του Παχέος Εντέρου
| Τύπος Μετάλλαξης | Εμπλεκόμενα Γονίδια | Τύπος Νόσου |
| Γενετικός | APC*MMR** | Οικογενής Αδενωματώδης ΠολυποδίασηHNPCC*** (Σύνδρομο Lynch) |
| Σωματικός | Ογκογονίδιαmycrassrc |
erbB2
Ογκοκατασταλτικά
p53
DCC
APC
Γονίδια MMR
hMSH2
hMLH1
hPMS1
hPMS2
hMSH6
hMSH3Σποραδικός
*APC: Γονίδιο Αδενωματώδους Πολυποδιάσεως Παχέος Εντέρου
**MMR: Γονίδια επιδιόρθωσης αστόχου ζευγαρώματος
***HNPCC: Κληρονομούμενος Μη Πολυποδιασιακός Κολοορθικός Καρκίνος
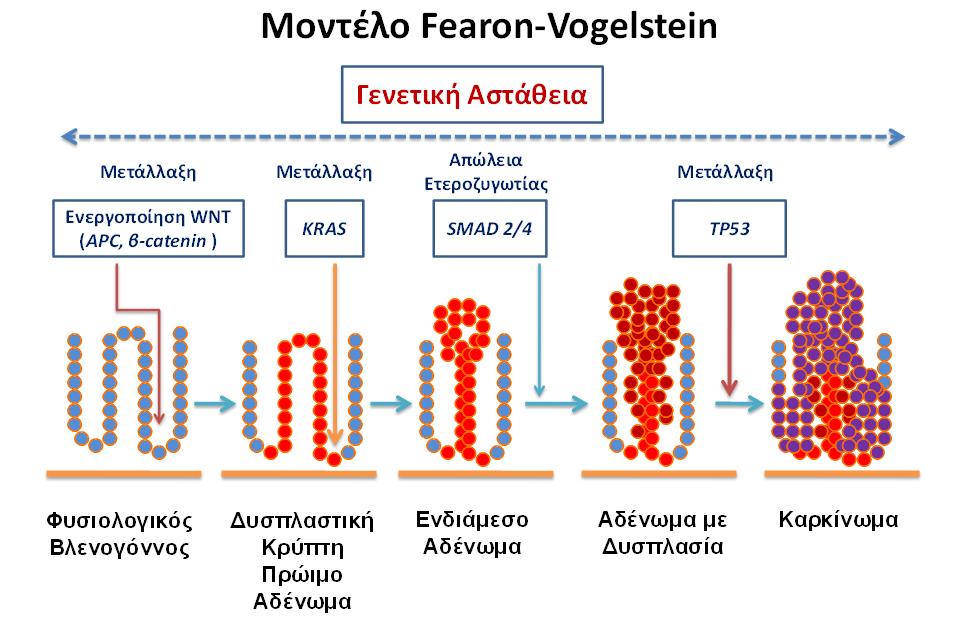
Εικόνα 2: Γενετικό πρότυπο της αναπτύξεως του καρκίνου του παχέος εντέρου.
Κατωτέρω περιγράφονται οι κύριες ανωμαλίες των ογκογονίδια, των ογκοκατασταλτικά γονίδια και των γονιδίων MMR, τα οποία αποτελούν την βάση της ογκογενέσεως του καρκίνου του παχέος εντέρου, συμπεριλαμβομένων της σπουδαιότητας κάθε παράγοντα στον κυτταρικό κύκλο και των μοριακών και κλινικών συνεπειών μεμονωμένων μεταλλάξεων. Αυτή η συζήτηση επιχειρεί να προσφέρει μία σύνοψη της μοριακής παθογενέσεως του κολοορθικού καρκίνου και όχι μία εξαντλητική ανασκόπηση. Μία περισσότερο σε βάθος ανάπτυξη διατίθεται αλλού10.
Ογκογονίδια – Τα ογκογονίδια είναι ομόλογα φυσιολογικών κυτταρικών γονιδίων τα οποία λαμβάνουν μέρος στις κυτταρικές οδούς αυξήσεως και ρυθμίσεως του κυτταρικού κύκλου. Μία αλλαγή εξ αιτίας μεταλλάξεως σε ένα ογκογονίδιο οδηγεί σε ενεργοποίηση του γονιδίου, με αποτέλεσμα τον ανεξέλεγκτο κυτταρικό πολλαπλασιασμό11,12. Μεταξύ των ογκογονιδίων τα οποία εμπλέκονται στον σποραδικό καρκίνο του παχέος εντέρου συγκαταλέγονται τα ras, c-myc και c-erbB-2 (HER2/neu). Το πιο σημαντικό από αυτά θεωρείται το ras (πίνακας 1)13-17.
Το ογκογονίδιο ras υφίσταται με την μορφή τριών μοριακών παραλλαγών: Η-ras, Κ-ras και Ν-ras. Παρ’ ότι και τα τρία ογκογονίδια μεταλλασσόμενα, έχουν την ικανότητα να οδηγούν σε εξαλλαγή τα φυσιολογικά κύτταρα, το Κ-ras είναι το συχνότερα μεταλλασσόμενο και υπεύθυνο για την ανάπτυξη του κολοορθικού καρκίνου στον άνθρωπο18-20. Η σπουδαιότητα του ras στη γένεση του καρκίνου του παχέος εντέρου υπογραμμίζεται από το εύρημα ότι τα καρκινικά κύτταρα, στα οποία ένα μεταλλαγμένο ras γονίδιο έχει αφαιρεθεί ή αντικατασταθεί, χάνουν την ικανότητα να σχηματίζουν όγκους σε ποντικούς.
Τα ογκογονίδια ras κωδικοποιούν μία οικογένεια μικρών πρωτεϊνών ομολόγων των G-πρωτεϊνών, οι οποίες ρυθμίζουν την μετάδοση μηνύματος στο κύτταρο ενεργώντας ως διακόπτης μιας κατευθύνσεως για την μετάδοση εξωκυτταρίων μηνυμάτων στον πυρήνα22. Αυτές οι πρωτεΐνες διαγράφουν κύκλους μεταξύ μιας ανενεργούς καταστάσεως δεσμευμένες με GDP και μιας ενεργούς καταστάσεως δεσμευμένες με GTP. Οι μεταλλάξεις του k-ras, κυρίως σημειακές, καθιστούν την πρωτεΐνη ανθεκτική στην υδρόλυση του GTP από την GTPάση, με αποτέλεσμα την συνεχή παρουσία ενεργούς πρωτεΐνης ras δεσμευμένης με GTP και κατά συνέπεια την ενός συνεχούς αυξητικού ερεθίσματος. Μετα-μεταφραστικές τροποποιήσεις της ras πρωτεΐνης από το ένζυμο φαρνεσύλ-τρανσφεράση είναι απαραίτητες για την τελική ενεργοποίησή της, και το γεγονός αυτό έχει αποτελέσει θεραπευτικό στόχο με την ανάπτυξη φαρμάκων τα οποία αναστέλλουν το συγκεκριμένο ένζυμο.
Μεταλλάξεις του ογκογονιδίου ras ανευρίσκονται σε ποσοστό έως και 50% των σποραδικών καρκίνων και στο 50% των αδενωμάτων μεγαλυτέρων του 1cm του παχέος εντέρου. Σπανίως παρατηρούνται σε μικρότερα αδενώματα18,23. Υποστηρίζεται ότι οι μεταλλάξεις του ογκογονιδίου ras είναι συνηθέστερες σε καρκίνους του ενδοπεριτοναϊκού κόλου σε σύγκριση με αυτούς του ορθού (61% έναντι 33% αντιστοίχως)24. Η απουσία μεταλλάξεων σε μικρότερα αδενώματα υποδηλώνει ότι οι μεταλλάξεις του ογκογονιδίου ras επισυμβαίνουν κατά την διάρκεια της ύστερης προόδου των αδενωμάτων25. Εντούτοις, οι μεταλλάξεις του ογκογονιδίου ras δεν περιορίζονται σε δυσπλαστικές αλλοιώσεις. Έως και το 100% των μη προδρόμων εστιακών δυσπλαστικών κρυπτών (aberrant crypt foci – ACF: ο πρώτος ενδιάμεσος σταθμός εξελίξεως μεταξύ της φυσιολογικής βλεννώδους μεμβράνης του εντέρου και του αδενωματώδους πολύποδα) και 25% των υπερπλασιακών πολυπόδων παρουσιάζουν μεταλλάξεις του ογκογονιδίου ras, αλλά η σημασία τους δεν είναι διευκρινισμένη18,21,22,26.
Η εξακρίβωση των μεταλλάξεων του ογκογονιδίου ras στον κολορθικό καρκίνο είναι δυνητικής κλινικής σημασίας στον προληπτικό έλεγχο και την θεραπεία. Η ανεύρεση μεταλλάξεων του ογκογονιδίου ras σε υλικό κοπράνων, ως ευαίσθητη μέθοδος ελέγχου για πρόωρη διάγνωση του καρκίνου του παχέος εντέρου, ευρίσκεται στο στάδιο της έρευνας27. Οι θεραπευτικές δυνατότητες παραγόντων οι οποίοι στοχεύουν την οδό επαγωγής μηνυμάτων του ογκογονιδίου ras (π.χ. αναστολείς της τρανσφεράσης της φαρνεστύλης), εξετάζεται σε ασθενείς με καρκίνο του παχέος εντέρου, των οποίων οι όγκοι περιέχουν μεταλλάξεις του ογκογονιδίου ras.
Ογκοκατασταλτικά Γονίδια – Σε αντίθεση με τα ογκογονίδια, τα ογκοκατασταλτικά γονίδια ασκούν φυσιολογικώς ανασταλτική επίδραση στον κυτταρικό κύκλο. Όταν αυτά τα γονίδια διαγραφούν ή η λειτουργία τους περιοριστεί, οι φυσιολογικοί μηχανισμοί ελέγχου καταστέλλονται και η κυτταρική αύξηση εξελίσσεται ανεξέλεγκτη. Στο κυτταρικό επίπεδο, η λειτουργία της φυσιολογικής πρωτεΐνης χάνεται μόνο όταν και τα δύο αλλήλια του γονιδίου είναι απενεργοποιημένα από σημειακές μεταλλάξεις, αναδιατάξεις ή διαγραφές.
Τα ογκοκατασταλτικά γονίδια περιγράφηκαν πρώτα από τον Knudson στο πλαίσιο του ρετινοβλαστώματος, το οποίο προκαλείται από απενεργοποίηση λόγω μεταλλάξεως του γονιδίου του ρετινοβλαστώματος (RB1), και παρουσιάζεται είτε ως κληρονομική είτε ως σποραδική ασθένεια28,29. Ένα πρότυπο “δύο κρούσεων” προτάθηκε για να εξηγήσει τα διαφορετικά κλινικά χαρακτηριστικά αυτών των δύο μορφών (εικόνα 3). Η κληρονομική μορφή του ρετινοβλαστώματος απαιτεί μία γαμετική μετάλλαξη η οποία είτε είναι κληρονομική είτε εμφανίζεται de novo (αποτέλεσμα νέας μεταλλάξεως μίας βλαστικής σειράς), με επιπλέον μία σωματική μετάλλαξη, η οποία θα επισυμβεί αργότερα κατά την ανάπτυξη και η οποία επηρεάζει το εναπομείναν RB1 αλλήλιο εντός των κυττάρων του αμφιβληστροειδούς12,28. Αυτές οι περιπτώσεις είναι κληρονομικές και οι προσβαλλόμενοι διατρέχουν τον κίνδυνο αναπτύξεως πολυεστιακών και αμφιπλεύρων όγκων. Αντιθέτως, στην μη κληρονομική μορφή του ρετινοβλαστώματος, οι μεταλλάξεις των δύο αλληλίων εμφανίζονται αυτομάτως σε ένα μόνο κύτταρο του αμφιβληστροειδούς (αμφότερες σωματικές μεταλλάξεις). Ο φαινότυπος ο οποίος προκύπτει είναι η ανάπτυξη ενός μονοεστιακού, μονοπλεύρου όγκου ο οποίος εμφανίζεται σε μεγαλύτερη ηλικία από ότι η κληρονομική παραλλαγή και χωρίς κληρονομήσιμη μετάδοση σε απογόνους29.
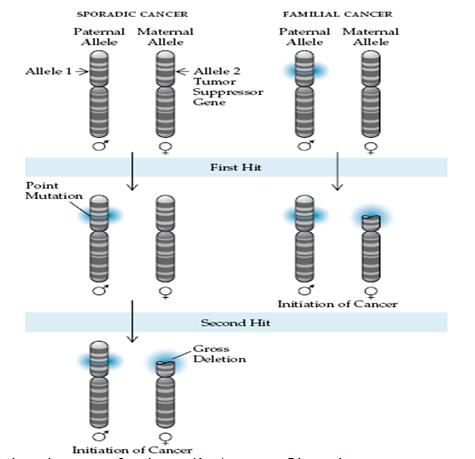
Εικόνα 3: Σχηματική παράσταση της θεωρίας του Knudston των «δύο κρούσεων».
Η πρώτη μοριακή απόδειξη για την εμπλοκή ογκοκατασταλτικών γονιδίων στην ανάπτυξη κολοορθικού καρκίνου προήλθε από την μελέτη της απώλειας των αλληλίων, όπου παρατηρήθηκαν μεγάλες χρωμοσωμικές διαγραφές με την χρήση πολυμορφικών γενετικών δεικτών, οι οποίοι διακρίνουν τα δύο αλλήλια παρόντα στη βλαστική σειρά. Σε σύγκριση μεταξύ των αλληλίων γονιδίου του όγκου και αλληλίων γονιδίων φυσιολογικών ιστών, εντοπίστηκαν διαγραφές με την μορφή της απωλείας της ετεροζυγωτίας (loss of heterozygosity – LOH) στην πρώτη περίπτωση.
Σε πρώιμες μελέτες καρκίνου του παχέος εντέρου, παρατηρήθηκε LOH στα χρωμοσώματα 5q, 8p, 17p ή 18q σε ποσοστά 36%, 50%, 73% και 75% αντιστοίχως23,30. Τα ογκοκατασταλτικά γονίδια του όγκου αναγνωρίστηκαν ακολούθως στο χρωμόσωμα 5q (γονίδια αδενωματώδους πολύποδα του κόλου ή APC γονίδια), στο χρωμόσωμα 18q (γονίδιο του καρκίνου του παχέος εντέρου [DCC gene: deleted colon cancer gene]) και τα SMAD4 και SMAD2 γονίδια και στο χρωμόσωμα 17p (p53 γονίδιο).
APC γονίδιο – Πιθανώς το πλέον κρίσιμο γονίδιο στην πρώιμη ανάπτυξη του καρκίνου του παχέος εντέρου είναι το ογκοκατασταλτικό APC γονίδιο. Σωματικές μεταλλάξεις και στα δύο αλλήλια γονίδια εμφανίζονται στο 80% των σποραδικών περιπτώσεων και μία μοναδική γαμετική μετάλλαξη σε αυτό το γονίδιο είναι υπεύθυνη για την ανάπτυξη ΣΟΑΠ. Η αναγνώριση της σπουδαιότητας του APC γονιδίου άρχισε με γονιδιακές μελέτες, οι οποίες συνδέουν την κληρονομικότητα του ΣΟΑΠ με το χρωμόσωμα 5q21, και τον επακόλουθο εντοπισμό μεταλλάξεων του βασικού γενετικού υλικού, στο οποίο περιλαμβάνεται ένα συγκεκριμένο γονίδιο σε αυτή την χρωμοσωμική θέση, το APC γονίδιο. Οι πλέον πρώιμες κακοήθεις αλλοιώσεις, όπως πρόδρομες εστιακές δυσπλαστικές κρύπτες – μικροαδενώματα και μικροί αδενωματώδεις πολύποδες, εμφανίζουν απώλεια και του δευτέρου APC αλληλόμορφου (διαμέσου διαγραφής ή σωματικής μεταλλάξεως), γεγονός το οποίο υποδηλώνει ότι η απώλεια του ογκοκατασταλτικού γονιδίου APC είναι ένα πολύ πρώιμο φαινόμενο στην ογκογένεση του παχέος εντέρου31,32.
Η λειτουργία του προϊόντος του γονιδίου APC και ο μηχανισμός δια του οποίου το μεταλλαγμένο γονίδιο προωθεί τον σχηματισμό όγκου έχουν αρχίσει ήδη να γίνονται κατανοητοί. Μία σημαντική ένδειξη υπήρξε η παρατήρηση ότι οι περισσότεροι σποραδικοί CRC με φυσιολογικό ή αγρίου τύπου APC παρουσίαζαν μεταλλάξεις σε β-κατενίνη, μία πρωτεΐνη η οποία εμπλέκεται στον ίδιο καταρράκτη μηνυμάτων όπως το APC, την οδό Wnt (Wingless-type)33,34. Η όδος Wnt αποτελεί μία εξελικτικώς διατηρουμένη οδό μεταβιβάσεως σημάτων, απαραιτήτων για την εμβρυϊκή ανάπτυξη34-36. Διαδραματίζει επίσης κεντρικό ρόλο στην ανανέωση του εντερικού επιθηλίου. Τα βασικά χαρακτηριστικά της οδού Wnt απεικονίζονται στην εικόνα 4. Η φυσιολογική APC πρωτεΐνη εμφανίζεται να παρεμποδίζει την συγκέντρωση κυτταροπλαστικής και πυρηνικής β-κατενίνης, μεσολαβώντας στη φωσφορυλίωση της και στην επερχόμενη αποδόμησή της. Η πλειοψηφία των μεταλλάξεων στο γονίδιο APC, τόσο των γαμετικών γραμμής όσο και των σωματικών, οδηγούν σε απώλεια της APC πρωτεΐνης και συνεπώς της ρυθμίσεως της β-κατενίνης, με αποτέλεσμα την συγκέντρωση β-κατενίνης στον πυρήνα, η οποία δεσμεύει και ενεργοποιεί τον παράγοντα μεταγραφής Τ-κυτταρικό παράγοντα-4 (T-cell factor – Tfc)37-39.
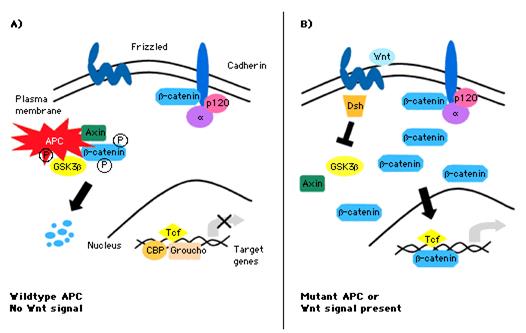
Εικόνα 4: Σχηματική παράσταση της απωλείας της λειτουργίας του APC και της οδού Wnt.
Υποστηρίζεται ότι η β-κατενίνη/Tfc-4 ενεργεί ως διακόπτης, ο οποίος ελέγχει τον πολλαπλασιασμό έναντι της διαφοροποιήσεως των εντερικών επιθηλιακών κυττάρων των κρυπτών40,41. Κατά συνέπεια η ενεργοποίηση της συγκεκριμένης οδού οδηγεί τα κύτταρα από την φάση G1 στη φάση S του κυτταρικού κύκλου, παρακάμπτοντας την φάση ελέγχου κατά το τέλος της φάσεως G1 και προκαλώντας αντίσταση στην απόπτωση των κυττάρων42. Το τελικό αποτέλεσμα είναι ο συνεχής κυτταρικός πολλαπλασιασμός. Επιπλέον, επειδή και άλλες κυτταρικές οδοί μηνυμάτων συγκλίνουν προς την οδό Wnt, αυτή αντιπροσωπεύει μία “τελική κοινή οδό” διαμέσου της οποίας πολλαπλές ανωμαλίες άλλων κυτταρικών οδών μηνυμάτων καταλήγουν δυνητικώς στο ίδιο καρκινογενετικό αποτέλεσμα. Τέλος, επιπλέον μηχανισμοί δυνατόν να συμβάλλουν στο ογκογενέτικο δυναμικό των μεταλλάξεων του APC. Μεταλλάξεις στο APC (άλλα όχι στη β-κατενίνη) συνδέονται με χρωμοσωμική αστάθεια43, καθιστώντας το κύτταρο ευένδοτο στην δράση και άλλων γονιδίων, ικανών να συνεισφέρουν στην πρόοδο του όγκου και την κακοήθη εξαλλαγή.
APC γονίδιο και οικογενής καρκίνος παχέος εντέρου – Σε Εβραίους Ashkenazi, ο οικογενής καρκίνος του παχέος εντέρου έχει συνδεθεί με μετάλλαξη του βασικού γενετικού υλικού στο κωδικόνιο 1307 (το οποίο περιέχει τα νουκλεοτίδια 3919, 3920 και 3921) του γονιδίου APC, γνωστή ως η μετάλλαξη Ι1307Κ του APC12,44,45. Η μετάλλαξη παρατηρείται στο 6% όλων των ατόμων με πρόγονο Εβραίο Ashkenazi και στο 28% των Εβραίων Ashkenazi, οι οποίοι έχουν προσωπικό ή και οικογενειακό ιστορικό κολοορθικού καρκίνου44. Αυτή η μετάλλαξη αρχικώς θεωρήθηκε ότι αντιπροσώπευε έναν πολυμορφισμό. Ο σχετικός κίνδυνος για ανάπτυξη καρκίνου του παχέος εντέρου στα συγκεκριμένα άτομα, αν και υψηλός σε σχέση με τον γενικό πληθυσμό, είναι πολύ χαμηλότερος συγκρινόμενος με ασθενείς οι οποίοι πάσχουν από ΣΟΑΠ44.
γονίδιο p53 – Το γονίδιο p53 στο χρωμόσωμα 17p είναι το συνηθέστερο μεταλλαγμένο στον καρκίνο στον άνθρωπο. Οι αλληλουχίες του 17p έχουν απωλεσθεί στο 75% των κολοορθικό καρκίνο, ενώ σπανίως παρατηρείται απώλεια σε αδενώματα και πρόδρομες εστιακές δυσπλαστικές κρύπτες, γεγονός το οποίο υποδηλώνει ότι η απώλεια του γονιδίου p53 αναπαριστά ένα σχετικώς καθυστερημένο γεγονός στην ογκογένεση του παχέος εντέρου7,23,46.
Το φυσιολογικό (αγρίου τύπου-wild type) γονίδιο p53 κωδικοποιεί μία πρωτεΐνη η οποία δεσμεύει το DNA, δρώντας ως μεταγραφικός ενεργοποιητής των ανασταλτικών γονιδίων της αυξήσεως. Το αγρίου τύπου γονίδιο p53 είναι ιδιαιτέρως σημαντικό σε καταστάσεις κυτταρικής πιέσεως. Κανονικώς τα κύτταρα αναστέλλουν την αύξησή τους ως απάντηση σε παράγοντες οι οποίοι καταστρέφουν το DNA αλλά και σε άλλα ερεθίσματα, όπως η υποξία, μέσω επαγωγής-ενεργοποιήσεως του γονιδίου p5347-49. Αφού ενεργοποιηθεί, το γονίδιο p53 προκαλεί μία ποικιλία από δράσεις οι οποίες περιορίζουν την αύξηση, συμπεριλαμβανομένων της διακοπής του κυτταρικού κύκλου (για να διευκολυνθεί η διόρθωση του DNA), της αποπτώσεως, της γηράνσεως και της διαφοροποιήσεως. Το p53 προάγει αυτές τις απαντήσεις σε μεγάλο βαθμό τροποποιώντας την έκφραση ενός αριθμού γονιδίων-στόχων, εκ των οποίων είκοσι τουλάχιστον έχουν περιγραφεί ως ευρισκόμενα κάτω από τον μεταγραφικό έλεγχο του p53. Εξ αιτίας του κεντρικού του ρόλου στην πρόληψη του πολλαπλασιασμού των κυττάρων με βλάβη στο DNA, το p53 αναφέρεται ως “ο φύλακας του γονιδιώματος” (guardian of the genom)50.
Η απενεργοποίηση της οδού του p53 είναι ένα καθυστερημένο γεγονός στην πλειοψηφία των κολοορθικών καρκίνων, αν και μπορεί να αντιπροσωπεύει ένα πρώιμο γεγονός στον καρκίνο του παχέος εντέρου ο οποίος σχετίζεται με φλεγμονώδη ασθένεια του εντέρου. Στο 50% με 70% των ασθενών με καρκίνο του παχέος εντέρου, η απενεργοποίηση του p53 συμβαίνει από μία μετάλλαξη του ενός αλληλομόρφου γονιδίου, η οποία ακολουθείται από την απώλεια του εναπομένοντος γονιδίου αγρίου τύπου23,51-53. Δεδομένης της σχέσεως της απενεργοποιήσεως της οδού του γονιδίου p53 με τον κολοορθικό καρκίνο, παραμένει ασαφές γιατί ασθενείς με το σύνδρομο Li Fraumeni, μία κατάσταση η οποία προκαλείται από μία γαμετική μετάλλαξη του p53 και κατά την οποία οι ασθενείς συχνά αναπτύσσουν καρκινώματα, σαρκώματα και λευχαιμία, δεν παρουσιάζουν ιδιαιτέρως υψηλή επικινδυνότητα αναπτύξεως καρκίνου του παχέος εντέρου.
Ο εντοπισμός μεταλλάξεων του p53 σε ένα μεμονωμένο κολοορθικό καρκίνο έχει δυνητική κλινική σημασία, σε επίπεδο προγνώσεως και θεραπείας. Σε πολλές, αλλά όχι σε όλες τις μελέτες, ασθενείς των οποίων οι όγκοι παρουσιάζουν μεταλλάξεις του p53 έχουν χειρότερα αποτελέσματα και μικρότερη επιβίωση από αυτούς χωρίς τις συγκεκριμένες μεταλλάξεις. Η προγνωστική σημασία των μεταλλάξεων του p53 σε ασθενείς με χειρουργημένο καρκίνο του παχέος εντέρου αποτελεί πεδίο έντονης ερευνητικής δραστηριότητας54-57.
Χρωμόσωμα 18q: Γονίδια DCC, SMAD4 και SMAD2 – Όπως στις περιπτώσεις των γονιδίων p53 και APC, το πρώτο πειστήριο για ένα ογκοκατασταλτικό γονίδιο στο χρωμόσωμα 18q προήλθε από μελέτες απωλειών αλληλίων σε περιπτώσεις κολοορθικού καρκίνου. Σε μία από τις πρώτες μελέτες, απώλεια ετεροζυγωτίας (LOH) του 18q παρατηρήθηκε στο 73% του σποραδικού καρκίνου του παχέος εντέρου και στο 47% των μεγάλων αδενωμάτων με εστίες διηθητικού καρκίνου, αλλά σε λιγότερο από 15% των πρωϊμων αδενωμάτων23. Το 1989 εντοπίστηκε ένα υποψήφιο γονίδιο επονομαζόμενο το “διαγραμμένο στον καρκίνο του κόλου” (DCC: deleted in colon cancer) στο χρωμόσωμα 18q2158, επί του οποίου εντοπίστηκαν σημειακές μεταλλάξεις σε ασθενείς με καρκίνο του παχέος εντέρου59,60.
Οι μεταλλάξεις του ως άνω γονιδίου πιθανώς οδηγούν στην απώλεια εκφράσεως της πρωτεΐνης DCC, η οποία πιστεύεται ότι διαδραματίζει κάποιον ρόλο στις αλληλεπιδράσεις μεταξύ κυττάρων ή μεταξύ κυττάρου και στρώματος58,61,62. Το γονίδιο DCC φυσιολογικώς εκφράζεται σε πολλούς ιστούς, συμπεριλαμβανομένου και του βλεννογόνου του παχέος εντέρου, αν και η φυσιολογική λειτουργία του είναι δύσκολο να αποσαφηνιστεί, εξ αιτίας του μεγάλου μεγέθους του και της απουσίας εκφράσεώς του σε πολλούς κολοορθικούς καρκίνους61.
Η απώλεια εκφράσεως του DCC δυνατό να έχει προγνωστική αξία, ειδικότερα σε ασθενείς με κολοορθικό καρκίνο πρωϊμου σταδίου. Το ποσοστό πενταετούς επιβιώσεως είναι χαμηλότερο σε ασθενείς με νόσο σταδίου ΙΙ στους οποίους απουσιάζει η έκφραση του γονιδίου DCC, σε σύγκριση με αυτούς οι οποίοι το εκφράζουν. Για τους ασθενείς με νόσο σταδίου ΙΙ και απουσία εκφράσεως του γονιδίου DCC, η πρόγνωση προσεγγίζει πολύ περισσότερο αυτή των ασθενών με νόσο σταδίου ΙΙΙ63. Το κέρδος από αυτή την πληροφορία είναι η δυνατότητα εντοπισμού ενός υποσυνόλου ασθενών με καρκίνο παχέος εντέρου σταδίου ΙΙ, οι οποίοι πιθανόν να οφελειθούν από συμπληρωματική χημειοθεραπεία. Αν και η ανωτέρω υπόθεση φαίνεται ελκυστική, δεν υπάρχουν επί του παρόντος δεδομένα, τα οποία υποστηρίζουν την εγκυρότητα αυτής της στρατηγικής.
Ένα δεύτερο ογκοκατασταλτικό γονίδιο εντοπίστηκε στο χρωμόσωμα 18q κατά την πορεία εξετάσεως της απώλειας ετεροζυγωτίας στον καρκίνο του παγκρέατος, το επονομαζόμενο DPC4 γονίδιο (deleted in pancreatic cancer – διαγραφόμενο στον καρκίνο του παγκρέατος – DPC), πλέον επαναπροσδιοριζόμενο ως SMAD423. Το SMAD4 κωδικοποιεί μία πρωτεΐνη η οποία δυνατό να είναι σημαντική στην οδό του μηνύματος της οικογενείας των πολυπεπτιδίων του νεοπλασματικού αυξητικού παράγοντα-βήτα (TGF-beta). Η πρωτεΐνη TGF-beta καταστέλλει την ανάπτυξη των περισσοτέρων φυσιολογικών κυττάρων, ενώ πολλά καρκινικά κύτταρα είναι ανθεκτικά σε αυτή την καταστολή της αυξήσεως. Στα καρκινικά κύτταρα του κολικού επιθηλίου, το γονίδιο SMAD4 είναι απαραίτητο για την αποστολή μηνύματος μέσω της πρωτεΐνης TGF-beta και, τουλάχιστον in vitro, η επανεισαγωγή του γονιδίου, δια της επιμολύνσεως ενός αθίκτου χρωμοσώματος 18, συνδέεται με την αποκατάσταση της ευαισθησίας του καρκινικού κυττάρου στην πρωτεΐνη TGF-beta64,65.
Μεταλλάξεις στο γονίδιο SMAD4 ή σε ένα τρίτο υποτιθέμενο ογκοκατασταλτικό γονίδιο, το οποίο χαρτογραφείται στο 18q (SMAD2), έχει βρεθεί σε ένα υποσύνολο σποραδικών κολοορθικών καρκίνων66-69. Ίσως είναι σημαντικότερο το γεγονός ότι γαμετικές μεταλλάξεις στο γονίδιο SMAD4 και στο γονίδιο BMPR1A, ένα μέλος της οικογένειας του υποδοχέα του TGF-beta ευρισκόμενο ανιόντως του SMAD470, έχουν επίσης εντοπιστεί σε ασθενείς με νεανική πολυποδίαση. Αυτοί οι ασθενείς αναπτύσσουν πολλαπλούς νεανικούς πολύποδες, διακρινομένους από τα αδενώματα, και διατρέχουν αυξημένο κίνδυνο αναπτύξεως διηθητικού καρκίνου του παχέος εντέρου.
Γονίδια Επιδιορθώσεως DNA
Τα γονίδια επιδιορθώσεως του DNA (MMR: missmatch repair) είναι υπεύθυνα για την διόρθωση του αστόχου ζευγαρώματος των νουκλεοτιδικών βάσεων και των μικρών εισαγωγών ή διαγραφών, οι οποίες επισυμβαίνουν κατά τον διπλασιασμό του DNA71-74. Για τα περισσότερα γονίδια MMR, συμπεριλαμβανομένων των hMSH2 (human mutS homolog 2), hMLH1 (human mutL homolog 1), hPMS1 και hPMS2 (human postmeiotic segregation 1 and 2), hMSH6 (human mutS homolog 6) και hMLH3, υπάρχει ένα γονίδιο υπεύθυνο για την διόρθωση του αποτυχημένου ζευγαρώματος, το οποίο αλληλεπιδρά με τον παράγοντα MLH1.
Οι γαμετικές μεταλλάξεις σε ένα από τα γονίδια MMR φαίνεται να είναι η βασική γενετική διαταραχή στους περισσοτέρους συγγενείς με κληρονομικό καρκίνο παχέος εντέρου χωρίς πολυποδίαση (HNPCC: nonpolyposis colorectal cancer), ενώ ανωμαλίες των MMR γονιδίων ανευρίσκονται στο 15% με 20% των σποραδικών κολοορθικών καρκίνων75-77. Εντούτοις, σποραδικοί όγκοι με ελαττωματικά MMR δεν περιέχουν γονιδιακές μεταλλάξεις των MMR, αλλά φέρουν επιγενετικές αλλαγές, οι οποίες επιβάλλουν σιωπή στην γονιδιακή έκφραση.
Κύτταρα με ελαττωματικά MMR γονίδια συσσωρεύουν λάθη του DNA εντός του γονιδιώματος78. Ειδικότερα, το βιολογικό “ίχνος” ενός ελαττωματικού MMR συστήματος είναι η συσσώρευση ανωμαλιών σε βραχείες αλληλουχίες νουκλεοτιδικών βάσεων, οι οποίες επαναλαμβάνονται δεκάδες έως εκατοντάδες φορές μέσα στο γονιδίωμα. Οι συγκεκριμένες αλληλουχίες καλούνται μικροδορυφόροι78. Αρκετά κρίσιμα γονίδια για την ρύθμιση της αυξήσεως (ο υποδοχέας του νεοπλασματικού αυξητικού παράγοντα βήτα τύπου ΙΙ TGF-β, ο ΒΑΧ, ο υποδοχέας του αυξητικού παράγοντα ΙΙ, ο οποίος προσομοιάζει της ινσουλίνης ILF II) περιέχουν μικροδορυφόρους στην περιοχή του εκκινητή, οι οποίοι είναι επιρρεπείς σε μεταλλάξεις, όταν υπάρχουν ελαττωματικά γονίδια MMR. Αυτό το φαινόμενο καλείται αστάθεια του μικροδορυφορικού DNA (microsetellite instabitily – MSI)78-80.
Σε αντίθεση με τον σταθερό, ως προς τους μικροδορυφόρους, κολοορθικό καρκίνο, οι σποραδικοί όγκοι, οι οποίοι παρουσιάζουν MSI, αποτελούν το 15% του συνόλου των σποραδικών καρκίνων του παχέος εντέρου και του ορθού και έχουν ιδιαίτερα κλινικοπαθολογικά χαρακτηριστικά. Ειδικότερα, τείνουν να εμφανίζονται στο εγγύς παχύ έντερο, έχουν μεγαλύτερη βλεννώδη σύνθεση, περιέχουν λεμφοκυτταρική διήθηση και είναι συνήθως ανεπαρκώς διαφοροποιημένοι. Παρά το τελευταίο χαρακτηριστικό, η παρουσία MSI συνδέεται με μακρύτερη επιβίωση τόσο στην HNPCC και στις σποραδικές περιπτώσεις81-86. Η παρουσία ή μη MSI καθορίζει δύο διακριτές ομάδες όγκων:
- νεοπλάσματα με χρωμοσωμιακή αστάθεια τα οποία χαρακτηρίζονται από απώλεια ευπλοϊδίας, απώλεια ετεροζυγωτίας (17p, 18q, 8p, 22q), μεταλλάξεις ογκοκατατσαλτικών γονιδίων (p53 και APC) και φυσιολογικό MMR σύστημα και αποτελούν το 85% των περιπτώσεων σποραδικού κολοορθικού καρκίνου και
- νεοπλάσματα με γενετική αστάθεια, οι οποίοι χαρακτηρίζονται από αστάθεια του μικροδοριφορικού DNA, ευπλοϊδικότητα, μεταλλάξεις των γονιδίων TGBβ-RII, BAX, TCF4, Caspase 5, HIF1α, αλλά όχι των ογκοκατασταλτικών γονιδίων, ενώ εμφανίζουν μεταλλάξεις ογκογονιδίων (BRAF και PI3KCA) και αποτελούν το 15% των περιπτώσεων σποραδικού κολορθικού καρκίνου.
Επιγενείς μεταβολές οι οποίες επηρεάζουν τα γονίδα MMR – Όπως ήδη προαναφέρθηκε, οι μεταλλάξεις και οι απώλειες αλληλομόρφων γονιδίων σε ένα από τα γονίδια MMR ευθύνονται για τον φαινότυπο MSI στις περισσότερες περιπτώσεις του HNPCC. Αντιθέτως, μεθυλίωση της περιοχής του εκκινητή ορισμένων γονιδίων MMR ή και η απώλεια μεταγραφής πιστεύεται ότι αποτελούν την γενετική βάση του σποραδικού καρκίνου του παχέος εντέρου, ο οποίος εμφανίζει φαινότυπο MSI7187-92. Επιπλέον, επιγενής απενεργοποίηση του δευτέρου φυσιολογικού αλληλομόρφου διαδραματίζει πιθανότατα κάποιο ρόλο σε ασθενείς με HNPCC, στους οποίους το δεύτερο αλλήλιο πρέπει να απενεργοποιηθεί για να εμφανισθεί τελικώς η νόσος93,94. Η μεθυλίωση του DNA συμβαίνει κατ’ εξοχήν σε CpG δινουκλεοτίδια, τα οποία είναι παρόντα στους εκκινητές πολλών γονιδίων, συμπεριλαμβανομένου του MMR του γονιδίου hMLH1. Αν και το ερέθισμα το οποίο οδηγεί σε υπερμεθυλίωση παραμένει άγνωστο, η μεθυλιωμένη κυτοσίνη δεσμεύεται από μία οικογένεια πρωτεϊνών, γνωστών ως πρωτεΐνες οι οποίες δεσμεύουν περιοχές μεθυλ-CpG, και σχηματίζει ένα πολυπρωτεϊνικό σύμπλοκο, το οποίο αλλάζει την διάταξη της χρωματίνης και καθιστά αδύνατη την έκφραση των γονιδίων95-97. Αξίζει να αναφερθεί ότι η μεθυλίωση των γονιδίνων του MMR συστήματος και κυρίως της hMLH1 έχει προσφάτως συσχετιστεί με αυξημένη εμφάνιση μεταλλάξεων (V600E) του γονιδίου BRAF 98-99, τονίζοντας εμφατικώς τον ρόλο του μονοπατιού του γονιδίου kras στον σποραδικό καρκίνο του παχέος εντέρου, τόσο σε αυτόν με χρωμοσωμική αστάθεια όσο και σε αυτόν με αστάθεια των μικροδορυφόρων.
Άλλος μηχανισμός επιγενούς απενεργοποιήσεως των MMR είναι η απώλεια αποτυπώσεως (loss of imprinting- LOI). Η απώλεια αποτυπώσεως αναφέρεται στην επιλεκτική απώλεια εκφράσεως των γαμετικών γονιδίων, εξ αιτίας επιλεκτικής μεθυλιώσεως ενός από τα αλληλόμορφα. Ο μηχανισμός διατηρείται κατά την διάρκεια της αναπτύξεως για να καταστείλει την έκφραση του μητρικού ή πατρικού αντιγράφου ενός αλληλομόρφου. Η LOI του γονιδίου του αυξητικού παράγοντα ΙΙ (IGF-II), οποίος προσομοιάζει της ινσουλίνης, έχει συνδεθεί με την ανάπτυξη καρκίνου του παχέος εντέρου με μεταβολές των MMR91,92,100. Εν τούτοις, η LOI του γονιδίου IGF-II ευρίσκεται συχνότερα στον φυσιολογικό βλεννογόνο του παχέος εντέρου και στα λεμφοκύτταρα του περιφερικού αίματος ασθενών με κολοορθικό καρκίνο, σε σύγκριση με αυτούς οι οποίοι δεν νοσούν91,101. Αυτά τα ευρήματα οδηγούν στην υπόθεση, ότι η LOI του γονιδίου IGF-II αντιπροσωπεύει έναν παράγοντα κινδύνου αναπτύξεως καρκίνου του παχέος εντέρου, παρά ένα σωματικό ελάττωμα το οποίο αποτελεί βάση για ογκογένεση, γενικώς101.
Ελλείψεις MMR και Οικογενής Κολοορθικός Καρκίνος – Μία μικρή αναλογία ασθενών με πολλαπλά κολοορθικά αδενώματα και οικογενειακό ιστορικό καρκίνου του παχέος εντέρου παρουσιάζουν μεταλλάξεις του βασικού γενετικού υλικού στο επανορθωτικό γονιδιακό ομόλογο εξαγωγής βάσεων mutY (MYH: mutY homolog), μερικές φορές με συνύπαρξη σωματικών μεταλλάξεων του γονιδίου APC102-104. Αυτές οι μεταλλάξεις προδιαθέτουν στην ανάπτυξη ενός συνδρόμου κληρονομουμένων πολλαπλών αδενωμάτων του παχέος εντέρου και στον φαινότυπο της κλασικής αδενωματώδους πολυποδιάσεως. Σε μία σειρά 152 ασθενών με πολλαπλά αδενώματα, 7,5% από αυτούς χωρίς γαμετική μετάλλαξη του APC παρουσίαζαν δύο ξεχωριστές γαμετικές μεταλλάξεις του ΜΥΗ102. Αυτά τα ευρήματα αναμένεται να επηρεάσουν τις στρατηγικές ελέγχου ασθενών με υποψία ΣΟΑΠ, το οποίο στις περισσότερες περιπτώσεις κληρονομείται με το αυτοσωμικό επικρατούν πρότυπο.
Επίσης, είναι σημαντικό ότι ένας αυξανόμενος αριθμός μελετών δείχνει ότι οι μεταλλάξεις στο γονίδιο ΜΥΗ δυνατό να ευθύνονται για ένα σημαντικό ποσοστό οικογενών κακρκίνων του παχέος εντέρου, οι οποίοι αναπτύσσονται χωρίς την παρουσία ενός ισχυρώς κληρονομικού οικογενούς συνδρόμου4-6,104-106. Σε μία σειρά βασισμένη σε 1.238 ασθενείς με καρκίνο παχέος εντέρου και 1.255 υγιείς παρομοίου φύλου και ηλικίας, οι οποίοι παρακολουθήθηκαν επί τρία έτη, μελετήθηκε η παρουσία μεταλλάξεων του γονιδίου ΜΥΗ (Υ165C και G382D). Οι φορείς μεταλλάξεων ήταν στατιστικώς περισσότερο πιθανόν να αναπτύξουν καρκίνου του παχέος εντέρου και να έχουν συγγενείς πρώτου ή δευτέρου βαθμού με κολοορθικό καρκίνο5.
Γονίδια Μετατροπείς
Επιπροσθέτως, υπάρχουν ενδείξεις ότι σειρά γονιδίων μετατροπέων ενέχονται στην ανάπτυξη του κολοορθικού καρκίνου, αν και οι ακριβείς ρόλοι και μηχανισμοί ογκογενέσεως δεν έχουν πλήρως διευκρινισθείί.
COX-2 – Υπάρχουν ενδείξεις ότι η ασπιρίνη ασπιρίνης και οι άλλοι αναστολείς της κυκλοοξυγενάσης προστατεύουν το παχύ έντερο από την ανάπτυξη καρκίνου. Επιπλέον ένας μη ειδικός COX αναστολέας, η σοθλινδάκη, προκαλεί υποστροφή των πολυπόδων σε ασθενείς με ΣΟΑΠ. Ο μηχανισμός με τον οποίον μεσολαβούνται αυτά τα φαινόμενα υποστροφής δεν είναι πλήρως κατανοητός. Σε κάθε περίπτωση η ενεργοποίηση της COX-2, είναι ένα σταθερό εύρημα τόσο σε αδενώματα όσο και σε διηθητικούς καρκίνους του παχέος εντέρου.
Γονίδιο PPAR – Το γονίδιο PPAR (PPAR: peroxisome proliferator-activating receptor) έχει εμπλακεί στην καρκινογενέση του παχέος εντέρου. Το γονίδιο PPAR κωδικοποιεί μια οικογένεια πυρηνικών υποδοχέων, οι οποίοι λειτουργούν ως μεταγραφικοί ρυθμιστές των πρωτεϊνών που ελέγχουν τον μεταβολισμό των λιπιδίων και την αύξηση των κυττάρων. Η ενεργοποίηση αυτών των υποδοχέων αναστέλλει την αύξηση του κυττάρου και προωθεί την διαφοροποίηση σε μία ποικιλία τύπων επιθηλιακών κυττάρων, συμπεριλαμβανομένων και των κυττάρων του κολοορθικού καρκίνου107. Πρόδρομες μελέτες δείχνουν ότι το γονίδιο PPAR βρίσκεται κατιόντως του γονιδίου APC και δυνατό να εμπλέκεται στην οδό της COX-2108-111.
Μεταλλάξεις απώλειας λειτουργίας του γονιδίου PPAR έχουν περιγραφεί σε σποραδικούς κολοορθικούς καρκίνους112. Επιπλέον, υπάρχουν ενδείξεις ότι οι ανωμαλίες του γονιδίου PPAR ευθύνονται για την αυξημένη συχνότητα καρκίνου και αδενωμάτων του παχέος εντέρου σε ασθενείς με ακρομεγαλία.
Ρύθμιση Λειτουργίας του Γονιδιώματος
Εκτός από τις δομικές αλλαγές στο γονιδίωμα, η λειτουργία του και κυρίως η μεταγραφή του, και ως εκ τούτου η πρωτεϊνική σύνθεση, έχει αποτελέσει πεδίο εντατικότατης έρευνας κυρίως την τελευταία τριετία. Η μεθυλίωση των εκκινητών των γονιδίων η οποία αναφέρθηκε ανωτέρω αποτελεί μία από τις πλέον εκτεταμένως μελετημένες μεταβολές της λειτουργίας του γονιδιώματος το οποίο εμπλέκεται στην καρκινογένεση. Εκτός όμως από την μεθυλίωση και η απακετυλίωση των ιστονών (πρωτεϊνών οι οποίες συνδέονται με το DNA και το οργανώνουν σε νουκλεοσώματα, τα οποία σχηματίζουν την χρωματίνη) φαίνεται ότι αποτελεί βασικό μηχανισμό της ρυθμίσεως της μεταγραφικής λειτουργίας του γονιδιώματος. Η ρύθμιση της μεθυλιώσεως και της απακετυλιώσεως των ιστονών και κυρίως των ελεύθερων τελικών τους άκρων (histone tails) οδηγεί σε «ανοιχτές» δομές χρωματίνης, οι οποίες επιτρέπουν την μεταγραφή του γονιδίου, ή σε κλειστές δομές χρωματίνης, οι οποίες οδηγούν σε σίγαση (εικόνα 5)113,114. Επίσης, προσφάτως πειραματικά δεδομένα υποστηρίζουν την ύπαρξη κώδικα ιστονών, ο οποίος καθορίζει τις βιοχημικές μεταβολές των τελικών άκρων τους και οδηγεί σε ανοιχτή ή κλειστή δομή χρωματίνης. (όπως ακριβώς ο κώδικας των νουκλεοτιδικών βάσεων στο mRNA καθορίζει την μετάφραση σε συγκεκριμένο αμινοξύ)115. Η διαταραχή των βιοχημικών μεταβολών των ιστονών στα καρκινικά κύτταρα συνιστά ένα από τα πλέον ενδιαφέροντα ερευνητικά πεδία σήμερα.
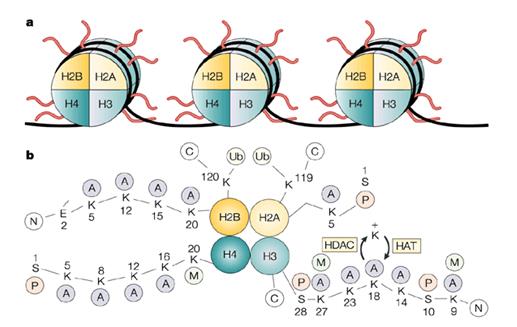
Εικόνα 5: Ιστόνες και δομή χρωματίνης. Βιοχημικές μετατροπές οι οποίες οδηγούν σε «ανοιχτή» δομή χρωματίνης.
Αυτό όμως το οποίο αποτέλεσε την πλέον σημαντική εξέλιξη στην έρευνα για την λειτουργία του γονιδιώματος είναι η ανεύρεση του ρόλου των micro RNAs. Τα micro RNAs (miRNA) είναι μικρά RNA (το πολύ έως 22 βάσεις), τα οποία δεν κωδικοποιούν πρωτεΐνες, αλλά ρυθμίζουν την έκφραση διαφόρων γονιδίων μέσω αναστολής της μεταφράσεως του αγγελιοφόρου RNA (εικόνα 6)116. Προέρχονται από φυλογενετικά διατηρημένες περιοχές του γονιδιώματος (από το ζυμομήκυτα έως τον άνθρωπο), γεγονός το οποίο υπογραμμίζει την σημασία τους στην ρύθμιση της λειτουργίας του γονιδιώματος. Έχει παρατηρηθεί ότι διαταραχές στην έκφραση των miRNAs είναι συχνή διαταραχή σε πολλές ανθρώπινες νεοπλασίες. Για παράδειγμα, η μείωση ή η εξάλειψη του miRNA let-7, το οποίο καταστέλλει την έκφραση του kras, οδηγεί σε υπερέκφραση της πρωτεΐνης και συνεχή ενεργοποίηση του μονοπατιού του γονιδίου kras χωρίς να υπάρχουν μεταλλάξεις στο γονίδιο117. Με αυτόν τον τρόπο τα miRNAs είναι σε θέση τα ίδια να διαδραματίσουν ρόλο ογκογονιδίων, καταστέλλοντας την έκφραση ογκοκατασταλτικών γονιδίων.
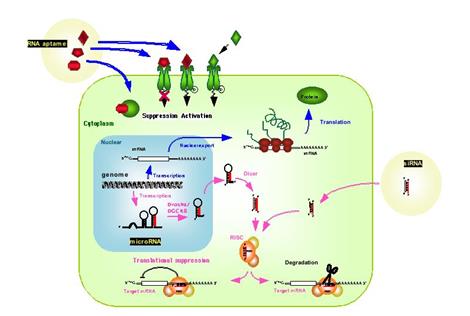
Εικόνα 6: Βιοσύνθεση και τρόπος δράσης των miRNAs.
ΠΕΡΙΛΗΨΗ: ΤΑ ΠΟΛΛΑΠΛΑ ΒΗΜΑΤΑ ΤΗΣ ΔΙΑΔΙΚΑΣΙΑΣ ΚΑΡΚΙΝΟΓΕΝΕΣΕΩΣ
Ο κολοορθικός καρκίνος αντιπροσωπεύει ένα ιδανικό πρότυπο μελέτης της μοριακής παθογενέσεως του καρκίνου, εξ αιτίας της προσβασιμότητας στον ιστό για βιοψία και της καθαρής εξελίξεως από φυσιολογικό επιθήλιο στο πρόδρομο στάδιο του αδενωματώδους πολύποδα και ακολούθως στον διεισδυτικό καρκίνο. Μία διαδικασία πολλαπλών βημάτων ειδικών γενετικών αλλαγών πιστεύεται ότι οδηγεί στον ανωτέρω μετασχηματισμό. Οι μεταλλάξεις του βασικού γενετικού υλικού αποτελούν την βάση για κληρονομικά σύνδρομα (ΣΟΑΠ, HNPCC), ενώ σποραδικοί καρκίνοι προκύπτουν από συσσώρευση πολλαπλών σωματικών μεταλλάξεων. Οι μεταλλάξεις στο γονίδιο APC λαμβάνουν χώρα στα αρχικά στάδια της διαδικασίας, ενώ άλλες, όπως οι μεταλλάξεις του κατασταλτικού ογκογονιδίου p53, επισυμβαίνουν στα τελικά στάδια (εικόνα 2)78.
Η έννοια του MSI παρέχει ένα επιπλέον επίπεδο πολυπλοκότητας σε αυτό το πρότυπο. Τα διαθέσιμα δεδομένα δείχνουν ότι οι περισσότερες νεοπλασίες του παχέος εντέρου αρχίζουν με την απενεργοποίηση του γονιδίου APC, ασχέτως της παρουσίας ή μη MSI. Εν τούτοις, τα γενετικά γεγονότα δυνατό να αποκλείνουν σε απώτερο στάδιο, εξαρτώμενα από τον μηχανισμό στον οποίο βασίζεται η γενετική αστάθεια (χρωμοσωμιακή αστάθεια ή MSI). Συγκρινόμενο με την χρωμοσωμιακή αστάθεια, η οποία αποτελεί το παθογενετικό υπόστρωμα της οικογενούς αδενωματώδους πολυποδιάσεως και της πλειοψηφίας των σποραδικών καρκίνων του παχέος εντέρου, η MSI ή ο “μεταλλάκτης” της οδού εμπλέκει έναν μοναδικό αποσταθεροποιητικό μηχανισμό και μία απενεργοποίηση διαφορετικού υποσυνόλου γονιδίων. Όμως, ανεξαρτήτως της εμπλεκομένης μοριακής οδού, το αποτέλεσμα είναι το ίδιο, η ανάπτυξη καρκίνου του παχέος εντέρου. Σε κάθε περίπτωση, στον ορθοκολικό καρκίνο η ενεργοποίηση του kras μονοπατιού φαίνεται να είναι κομβικό γεγονός για την συντήρηση της νεοπλασίας και την εξέλιξη της σε πλέον διηθητικές μορφές.
Επιπλέον, ο εντοπισμός συγκεκριμένων γενετικών μεταλλάξεων υπευθύνων για την ογκογένεση του παχέος εντέρου έχει άμεση επίδραση στην κλινική πράξη. Οι ασθενείς με υψηλότερο κίνδυνο εμφανίσεως κολορθικού καρκίνου εντοπίζονται μέσω γενετικών ελέγχων για συγκεκριμένες μεταλλάξεις του γενετικού υλικού. Νέες μοριακές μέθοδοι εξετάσεως για πρώιμη ανεύρεση του καρκίνου του παχέος εντέρου, με εντοπισμό μεταλλάξεων σε υλικό κοπράνων ευρίσκονται ήδη στο στάδιο μελέτης. Επιπλέον, αυτές οι μεταλλάξεις εξετάζονται επίσης ως προγνωστικοί δείκτες, αλλά και ως δυνητικοί θεραπευτικοί στόχοι.
Τέλος, η ρύθμιση της λειτουργίας του γονιδιώματος, είτε μέσω μεθυλιώσεως των εκκινητών, είτε μέσω βιοχημικών μετατροπών των τελικών αμινοξέων των ιστονών, είτε μέσω των miRNA, έχει ουσιαστικό ρόλο στην εξέλιξη της νεοπλασίας και αποτελεί δυνητικό θεραπευτικό στόχο.
ΒΙΒΛΙΟΓΡΑΦΙΑ
- Jemal A, Murray T, Ward W, et al: Cancer statistics, 2005. CA Cancer J Clin 2005; 55: 10-30.
- Wirtzfeld DA, Petrelli NJ, Rodriguez-Bigas MA: Hamartomatous polyposis syndromes: molecular genetics, neoplastic risk, and surveillance recommendations. Ann Surg Oncol 2001; 8: 319-27.
- Pinsky PF: Does hereditary nonpolyposis colorectal cancer explain the observed excess risk of colorectal cancer associated with family history? Epidemiology 2000; 11: 297-303.
- Ricciardiello L, Goel A, Mantovani V, et al: Frequent loss of hMLH1 by promoter hypermethylation leads to microsatellite instability in adenomatous polyps of patients with a single first-degree member affected by colon cancer. Cancer Res 2003; 63: 787-92.
- Croitoru ME, Cleary SP, Di Nicola N, et al: Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst 2004; 96: 1631-4.
- Enholm S, Hienonen T, Suomalainen A, et al: Proportion and phenotype of MYH-associated colorectal neoplasia in a population-based series of Finnish colorectal cancer patients. Am J Pathol 2003; 163: 827-32.
- Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759-67.
- Winawer SJ, Zauber AG, Ho MN, et al: Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp Study Work Group. N Engl J Med 1993; 329: 1977-81.
- Das PM, Singal R: DNA methylation and cancer. J Clin Oncol 2004; 22: 4632-42.
- Lynch JP, Hoops TC: The genetic pathogenesis of colorectal cancer. Hematol Oncol Clin North Am 2002; 16: 775-810.
- Sherr CJ: Cancer cell cycles. Science 1996; 274: 1672-7.
- Cancer Genetics and Cancer Predisposition Testing. American Society of Clinical Oncology Curriculum. Alexandria, VA: American Soc of Clinical Oncology; 1998.
- Forgacs I: Oncogenes and gastrointestinal cancer. Gut 1988; 29: 417-21.
- Cartwright C: Intestinal cell growth control: Role of Src tyrosine kinases (editorial). Gastroenterology 1998; 114: 1335-8.
- Hamilton SR: The molecular genetics of colorectal neoplasia. Gastroenterology 1993; 105: 3-7.
- Kapitanovic S, Radosevic S, Kapitanovic M et al: The expression of p185(HER-2/neu) correlates with the stage of disease and survival in colorectal cancer. Gastroenterology 1997; 112: 1103-13.
- Irby RB, Mao, W Coppola D et al: Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet 1999; 21: 187-90.
- Takayama T, Ohi M, Hayashi T et al: Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology 2001; 121: 599-611.
- Shibata D, Schaeffer J, Li ZH et al: Genetic heterogeneity of the c-K-ras locus in colorectal adenomas but not in adenocarcinomas. J Natl Cancer Inst 1993; 85: 1058-63.
- Tortola S, Marcuello E, Gonzalez I et al: p53 and K-ras gene mutations correlate with tumor aggressiveness but are not of routine prognostic value in colorectal cancer. J Clin Oncol 1999; 17: 1375-81.
- Shirasawa S, Furuse M, Yokoyama N, Sasazuki T: Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 1993; 260: 85-8.
- Bourne HR, Sanders DA, McCormick F: The GTPase superfamily: conserved structure and molecular mechanism. Nature 1991; 349: 117-27.
- Vogelstein B, Fearon ER, Hamilton SR et al: Genetic alterations during colorectal-tumor development. N Engl J Med 1988; 319: 525-32.
- Frattini M, Balestra D, Suardi S et al: Different genetic features associated with colon and rectal carcinogenesis. Clin Cancer Res 2004; 10: 4015-21.
- Pretlow TP, Brasitus TA, Fulton NC et al: K-ras mutations in putative preneoplastic lesions in human colon. J Natl Cancer Inst 1993; 85: 2004-7.
- Losi L, Roncucci L, di Gregorio C et al: K-ras and p53 mutations in human colorectal aberrant crypt foci. J Pathol 1996; 178: 259-63.
- Ahlquist DA, Skoletsky JE, Boynton KA et al: Colorectal cancer screening by detection of altered human DNA in stool: feasibility of a multitarget assay panel. Gastroenterology 2000; 119: 1219-27.
- Knudson AG Jr: Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971; 68: 820-3.
- Knudson AG Jr: Hereditary cancer, oncogenes, and antioncogenes. Cancer Res 1985; 45: 1437-43.
- Vogelstein B: Allelotype of colorectal carcinomas. Science 1989; 244: 207-11.
- Spirio LN, Samowitz W, Robertson J et al: Alleles of APC modulate the frequency and classes of mutations that lead to colon polyps. Nat Genet 1998; 20: 385-8.
- Lamlum H, Ilyas M, Rowan A et al: The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: A new facet to Knudson’s ‘two-hit’ hypothesis. Nat Med 1999; 5: 1071-5.
- Su LK, Vogelstein B, Kinzler KW: Association of the APC tumor suppressor protein with catenins. Science 1993; 262: 1734-7.
- Bienz M, Clevers H: Linking colorectal cancer to Wnt signaling. Cell 2000; 103: 311-20.
- Fearnhead NS, Britton MP, Bodmer W: The ABC of APC. Hum Mol Genet 2001; 10: 721-33.
- Uthoff SM, Eichenberger MR, McAuliffe TL et al: Wingless-type frizzled protein receptor signaling and its putative role in human colon cancer. Mol Carcinog 2001; 31: 56-62.
- Korinek V, Barker N, Morin PJ et al: Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 1997; 275: 1784-7.
- Morin PJ, Sparks AB, Korinek V et al: Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997; 275: 1787-90.
- Goss KH, Groden J: Biology of the adenomatous polyposis coli tumor suppressor. J Clin Oncol 2000; 18: 1967-79.
- Van de Wetering M, Sancho E, Verweij C et al: The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002; 111: 241-50.
- Shih IM, Wang TL, Traverso G et al: Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci U S A 2001; 98: 2640-5.
- Kim PJ, Plescia J, Clevers H et al: Survivin and molecular pathogenesis of colorectal cancer. Lancet 2003; 362: 205-9.
- Fodde R, Kuipers J, Rosenberg C et al: Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol 2001; 3 : 433-8.
- Laken SJ, Petersen GM, Gruber SB et al: Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat Genet 1997; 17: 79-83.
- Drucker L, Shpilberg O, Neumann A et al: Adenomatous polyposis coli I1307K mutation in Jewish patients with different ethnicity: prevalence and phenotype. Cancer 2000; 88: 755-60.
- Kirsch DG, Kastan M: Tumor-suppressor p53: Implications for tumor development and prognosis. J Clin Oncol 1998; 16: 3158-68.
- Kastan MB, Onyekwere O, Sidransky D et al: Participation of p53 protein in the cellular response to DNA damage. Cancer Res 1991; 51: 6304-11.
- Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB: Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci U S A 1992; 89: 7491-5.
- Woods DB, Vousden KH: Regulation of p53 function. Exp Cell Res 2001; 264: 56-66.
- Lane DP: Cancer: p53, guardian of the genome. Nature 1992; 358: 15-6.
- Soussi T: The p53 tumor suppressor gene: from molecular biology to clinical investigation. Ann N Y Acad Sci 2000; 910: 121-37.
- Baker SJ, Preisinger AC, Jessup JM et al: p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res 1990; 50: 7717-22.
- Kikuchi-Yanoshita R, Konishi M, Ito S et al. Genetic changes of both p53 alleles associated with the conversion from colorectal adenoma to early carcinoma in familial adenomatous polyposis and non-familial adenomatous polyposis patients. Cancer Res 1992; 52: 3965-71.
- Hamid O, Varterasian ML, Sadler S et al: Phase II trial of Intravenous CI-1042 in Patients With Metastatic Colorectal Cancer. J Clin Oncol 2003; 21: 1498-504.
- Warren RS, Kirn DH: Liver-directed viral therapy for cancer p53-targeted adenoviruses and beyond. Surg Oncol Clin N Am 2002; 11: 571-88.
- Raj K, Ogston P, Beard P: Virus-mediated killing of cells that lack p53 activity. Nature 2001; 412: 914-7.
- Watanabe T, Sullenger BA: Induction of wild-type p53 activity in human cancer cells by ribozymes that repair mutant p53 transcripts. Proc Natl Acad Sci U S A 2000; 97: 8490-4.
- Fearon ER, Cho KR, Nigro JM et al: Identification of a chromosome 18q gene that is altered in colorectal cancers. Science 1990; 247: 49-56.
- Hedrick L, Cho KR, Fearon ER et al: The DCC gene product in cellular differentiation and colorectal tumorigenesis. Genes Dev 1994; 8: 1174-83.
- Chan SS, Zheng H, Su MW et al: UNC-40, a C. elegans homolog of DCC (Deleted in Colorectal Cancer), is required in motile cells responding to UNC-6 netrin cues. Cell 1996; 87: 187-95.
- Thiagalingam S, Lengauer C, Leach FS et al: Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet 1996; 13: 343-6.
- Cho KR, Oliner JD, Simons JW et al: The DCC gene: structural analysis and mutations in colorectal carcinomas. Genomics 1994; 19: 525-31.
- Sun XF, Rutten S, Zhang H, Nordenskjold B: Expression of the deleted in colorectal cancer gene is related to prognosis in DNA diploid and low proliferative colorectal adenocarcinoma. J Clin Oncol 1999; 17: 1745-50.
- Goyette MC, Cho K, Fasching CL et al: Progression of colorectal cancer is associated with multiple tumor suppressor gene defects but inhibition of tumorigenicity is accomplished by correction of any single defect via chromosome transfer. Mol Cell Biol 1992; 12: 1387-95.
- Reiss M, Santoro V, de Jonge RR, Vellucci VF: Transfer of chromosome 18 into human head and neck squamous carcinoma cells: evidence for tumor suppression by Smad4/DPC4. Cell Growth Differ 1997; 8: 407-15.
- Riggins GJ, Thiagalingam S, Rozenblum E et al: Mad-related genes in the human. Nat Genet 1996; 13: 347-9.
- Eppert K, Scherer SW, Ozcelik H et al: MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell 1996; 86: 543-52.
- Mac Grogan D, Pegram M, Slamon D, Bookstein R: Comparative mutational analysis of DPC4 (Smad4) in prostatic and colorectal carcinomas. Oncogene 1997; 15: 1111-4.
- Xie W, Rimm DL, Lin Y et al: Loss of Smad signaling in human colorectal cancer is associated with advanced disease and poor prognosis. Cancer J 2003; 9: 302-12.
- Zhou XP, Woodford-Richens K, Lehtonen R et al: Germline mutations in BMPR1A/ALK3 cause a subset of cases of juvenile polyposis syndrome and of Cowden and Bannayan-Riley-Ruvalcaba syndromes. Am J Hum Genet 2001; 69: 704-11.
- Chung DC, Rustgi AK: DNA mismatch repair and cancer. Gastroenterology 1995; 109: 1685-99.
- Papadopoulos N, Nicolaides NC, Liu B et al: Mutations of GTBP in genetically unstable cells. Science 1995; 268: 1915-7.
- Baker SM, Bronner CE, Zhang L et al: Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell 1995; 82: 309-19.
- Papadopoulos N, Nicolaides NC, Wei YF et al: Mutation of a mutL homolog in hereditary colon cancer. Science 1994; 263: 1625-9.
- Boland CR, Thibodeau SN, Hamilton SR et al: A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998; 58: 5248-57.
- Syngal S, Weeks JC, Schrag D et al: Benefits of colonoscopic surveillance and prophylactic colectomy in patients with hereditary nonpolyposis colorectal cancer mutations. Ann Intern Med 1998; 129: 787-96.
- Shibata D, Peinado MA, Ionov Y et al: Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat Genet 1994; 6: 273-81.
- Ionov Y, Peinado MA, Malkhosyan S et al: Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993; 363: 558-61.
- Markowitz S, Wang J, Myeroff L et al: Inactivation of the type II TGF- receptor in colon cancer cells with microsatellite instability. Science 1995; 268: 1336-8.
- Fujiwara T, Stolker JM, Watanabe T et al: Accumulated clonal genetic alterations in familial and sporadic colorectal carcinomas with widespread instability in microsatellite sequences. Am J Pathol 1998; 153: 1063-78.
- Hatch SB, Lightfoot HM Jr, Garwacki CP et al: Microsatellite instability testing in colorectal carcinoma: choice of markers affects sensitivity of detection of mismatch repair-deficient tumors. Clin Cancer Res 2005; 11: 2180-7.
- Parc YR, Halling KC, Wang L et al: HMSH6 alterations in patients with microsatellite instability-low colorectal cancer. Cancer Res 2000; 60: 2225-31.
- Thibodeau SN, French AJ, Roche PC et al: Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res 1996; 56: 4836-40.
- Thibodeau SN, French AJ, Cunningham JM et al: Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Res 1998; 58: 1713-8.
- Laiho P, Launonen V, Lahermo P et al: Low-level microsatellite instability in most colorectal carcinomas. Cancer Res 2002; 62: 1166-70.
- Ashktorab H, Smoot DT, Carethers JM et al: High incidence of microsatellite instability in colorectal cancer from African Americans. Clin Cancer Res 2003; 9: 1112-7.
- Weinberg RA: Oncogenes and tumor suppressor genes. CA Cancer J Clin 1994; 44: 160-70.
- Veigl ML, Kasturi L, Olechnowicz J et al: Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A 1998; 95: 8698-702.
- Herman JG, Umar A, Polyak K et al: Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A 1998; 95: 6870-5.
- Cunningham JM, Kim CY, Christensen ER et al: The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas. Am J Hum Genet 2001; 69: 780-90.
- Cui H, Horon IL, Ohlsson R et al: Loss of imprinting in normal tissue of colorectal cancer patients with microsatellite instability. Nat Med 1998; 4: 1276-80.
- Nakagawa H, Chadwick RB, Peltomaki P et al: Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc Natl Acad Sci U S A 2001; 98: 591-6.
- Kane MF, Loda M, Gaida GM et al: Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997; 57: 808-11.
- Esteller M, Fraga MF, Guo M et al: DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet 2001; 10: 3001-7.
- Baylin SB, Esteller M, Rountree MR et al: Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet 2001; 10: 687-92.
- Tycko B: Epigenetic gene silencing in cancer. J Clin Invest 2000; 105: 401-7.
- Herman JG, Baylin SB: Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003; 349: 2042-54.
- Liang Wang, Cunningham J M, Winters J L et al: BRAF mutations in Colon Cancer are not likely attributed to defective DNA Mismatch Repair System 2003; Can Research: 5209-5212.
- Oliveira C, Pinto M, Duval A, et al: BRAF mutations characterize colon but not gastric cancer with mismatch repair deficiency. Oncogenes 2003; 22: 9191-9196.
- Rhee I, Bachman KE, Park BH et al: DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002; 416: 552-6.
- Cui H, Cruz-Correa M, Giardiello FM et al: Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science 2003; 299: 1753-5.
- Sieber OM, Lipton L, Crabtree M et al: Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med 2003; 348: 791-9.
- Sampson JR, Dolwani S, Jones S et al: Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003; 362: 39-41.
- Wang L, Baudhuin LM, Boardman LA et al: MYH mutations in patients with attenuated and classic polyposis and with young-onset colorectal cancer without polyps. Gastroenterology 2004; 127: 9-16.
- Kambara T, Whitehall VL, Spring KJ et al: Role of inherited defects of MYH in the development of sporadic colorectal cancer. Genes Chromosomes Cancer 2004; 40: 1-9.
- Al-Tassan N, Chmiel NH, Maynard J et al: Inherited variants of MYH associated with somatic G:C–>T: A mutations in colorectal tumors. Nat Genet 2002; 30: 227-32.
- Gupta RA, Brockman JA, Sarraf P et al: Target genes of peroxisome proliferator-activated receptor gamma in colorectal cancer cells. J Biol Chem 2001; 276: 29681-7.
- He TC, Chan TA, Vogelstein B, Kinzler KW: PPAR is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999; 99: 335-45.
- Yang WL, Frucht H: Activation of the PPAR pathway induces apoptosis and COX-2 inhibition in HT-29 human colon cancer cells. Carcinogenesis 2001; 22: 1379-83.
- Dobbie Z, Muller PY, Heinimann K et al: Expression of COX-2 and Wnt pathway genes in adenomas of familial adenomatous polyposis patients treated with meloxicam. Anticancer Res 2002; 22: 2215-20.
- Gupta, RA Tan J, Krause WF et al: Prostacyclin-mediated activation of peroxisome proliferator-activated receptor delta in colorectal cancer. Proc Natl Acad Sci USA 2000; 97: 13275-80.
- Sarraf P, Mueller E, Smith WM et al: Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell 1999; 3: 799-804.
- Marks PA, Rifkind RA, Richon VM, et al: Histone deacetylases and cancer: causes and therapies. Nat Ca Rev 2001; 194-202.
- Agger K, Christensen J, Cloos PA, et al: The emerging functions of histone demethylases. Curr Opin Genet Dev. 2008 Feb 15; [Epub ahead of print].
- Rathert P, Zhang X, Freund C, et al: Analysis of the substrate specificity of the dim-5 histone lysine methyltransferase using Peptide arrays. Chem Biol. 2008; 15: 5-11.
- Shivdasani R: MicroRNAs: Regulators of gene translation and cell differentiation. Blood 2006; 108: 3646-3653.
- Akao, Y., Nakagawa, Y., and Naoe, T. (2006). let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 29, 903–906.
Γενετική αστάθεια και τα μονοπάτια της καρκινογένεσης στον ορθοκολικό καρκίνο
Η συστηματική μελέτη των γενετικών και επιγενετικών μεταβολών που χαρακτηρίζουν τη νόσο υποδεικνύει ότι δε μπορούμε πλέον να αναφερόμαστε στον ΟΚΚ σαν ενιαία νόσο. Έχουν χαρακτηριστεί έως σήμερα τουλάχιστον 3 υποομάδες με διαφορετικούς μηχανισμούς έναρξης και ανάπτυξης της νεοπλασίας, οι οποίες συνοδεύονται με διακριτά κλινικο-εργαστηριακά χαρακτηριστικά και έχουν σημαντικές επιπτώσεις στην πρόληψη, τις στρατηγικές έγκαιρης διάγνωσης και το θεραπευτικό σχεδιασμό της νόσου (1).
Η γενετική αστάθεια είναι γνωστό ότι καθοδηγεί ποικίλες μοριακές μεταβολές που χαρακτηρίζουν την ανάπτυξη της νεοπλασίας και οι οποίες μέσω ενός Δαρβινικού μοντέλου επιλογής είναι υπεύθυνες για την έναρξη και την εξέλιξη της νεοπλασματικής μεταλλαγής. Ιδιαίτερα για τον ορθοκολικό καρκίνο είναι ξεκάθαρο ότι όλες οι περιπτώσεις χαρακτηρίζονται από γενετική αστάθεια, χάρις σε πειραματικά δεδομένα αναφοράς που προήλθαν κυρίως από το εργαστήριο του Bert Vogelstein (2). Η γενετική αυτή αστάθεια στην πλειονότητα (≈85-90%) των περιπτώσεων αφορά σε απώλεια μεγάλων τμημάτων κάποιων χρωμοσωμάτων (φαινόμενο το οποίο είναι γνωστό ως χρωμοσωμική αστάθεια-chromosomal instability, CIN) μέσω αγνώστου εν πολλοίς μηχανισμού, ενώ στις υπόλοιπες (≈10-15%) περιπτώσεις αφορά σε αστάθεια των μικροδορυφόρων του DNA (που καλείται συνήθως μικροδορυφορική αστάθεια-microsatelite instability, MSI) και οφείλεται στην απώλεια λειτουργίας του mismatch repair (MMR) συστήματος (3;4). Αυτό το γεγονός οδήγησε στο απλουστευμένο μοντέλο των δύο μονοπατιών που οδηγούν στην ανάπτυξη του ορθοκολικού καρκίνου: α) το μονοπάτι της χρωμοσωμικής αστάθειας (CIN) με το μοντέλο της εξέλιξης από αδένωμα σε καρκίνωμα (γνωστό και ως “Volgegram”) (5) και β) το μονοπάτι της μικροδορυφορικής αστάθειας (MSI-H) (6).
Η εικόνα, όμως, έγινε περισσότερο περίπλοκη όταν αναφέρθηκαν δεδομένα τα οποία έδειχναν ότι μερικές περιπτώσεις ορθοκολικού καρκίνου δεν μπορούσαν να ενταχθούν ούτε στο μονοπάτι της CIN ούτε σε αυτό του MSI-H (7). Παράλληλα, επιγενετικές αλλαγές κυρίως λόγω μεθυλίωσης του DNA αναγνωρίζονται όλο και συχνότερα σαν συχνό σύμβαμα στον ορθοκολικό καρκίνο και καθορίστηκε ένας υπότυπος της νόσου με σημαντική υπερμεθυλίωση ο ποίος χαρακτηρίστηκε σαν CIMP (CpG Island Methylator Phenotype) (8). Δεδομένης της λειτουργικής ισοδυναμίας μεταξύ της επιγενούς σίγασης και των αδρανοποιών μεταλλάξεων (9), η «επιγενετική αστάθεια» αποτελεί μία εναλλακτικό οδό για την ανάπτυξη του ορθοκολικού καρκίνου. Επιπροσθέτως, το θέμα περιπλέκεται περαιτέρω με το εύρημα ότι οι περισσότερες περιπτώσεις MSI στον σποραδικό ορθοκολικό καρκίνο οφείλονται σε CIMP-σίγαση του γονιδίου MLH1 (10) που κωδικοποιεί την αντίστοιχη πρωτεΐνη με κομβικό ρόλο στο MMR σύστημα. Νεότερα δεδομένα δείχνουν μία αντίστροφη συσχέτιση των δύο μηχανισμών οι οποίοι πολύ σπάνια, εάν ποτέ, αλληλεπικαλύπτονται (11;12). Αυτή η αντίθετη συσχέτιση οδηγεί σε μία ακόμη ένδειξη για την λειτουργική ισοδυναμία των γενετικών και επιγενετικών μηχανισμών στο να μεταβάλουν σημαντικά μονοπάτια στη διαδικασία ανάπτυξης του ορθοκολικού καρκίνου (13).
Είναι γνωστό ότι οι γενετικές μεταλλαγές που χαρακτηρίζουν τον ορθοκολικό καρκίνο, είναι πολύ καλά τεκμηριωμένες και οι συχνότερες από αυτές είναι οι μεταλλάξεις των APC, KRAS και TP53 ακολουθούμενες σε συχνότητα από τις μεταλλαγές των PIK3CA και BRAF (14). Παρ’ όλα υπάρχει μεγάλη ετερογένεια ανάλογα με το μηχανισμό που είναι υπεύθυνος για τη νεοπλασία. Οι περιπτώσεις με CIMP χαρακτηρίζονται κυρίως από μεταλλαγές του BRAF (έως και 50% των περιπτώσεων) και πολύ σπάνια των APC, KRAS και TP53. Αντιθέτως, οι όγκοι με CIN φέρουν στο σύνολο τους μία γενετική μεταβολή που οδηγεί σε ενεργοποίηση του μονοπατιού του Wnt (συνηθέστερα αδρανοποιό μετάλλαξη του APC) και πολύ συχνά μεταλλάξεις των KRAS και TP53 ενώ η μετάλλαξη του BRAF είναι εξαιρετικώς σπάνιο σύμβαμα (< 2%). Όπως έχει δειχθεί οι μεταλλάξεις που παρατηρούνται στον πρωτοπαθή όγκο ανευρίσκονται κατά κανόνα και στην μετάσταση (15;16). Παρ’ όλα αυτά πρόσφατα δεδομένα υποδηλώνουν την ύπαρξη ενός τρίτου πιθανού μονοπατιού για την ανάπτυξη ορθοκολικού καρκίνου (17). Το τρίτο αυτό μονοπάτι χαρακτηρίζεται από διαφορετικά CIMP πρότυπα φέρει συχνά μεταλλαγές του KRAS και ενίοτε του BRAF αλλά σχεδόν δε ανευρίσκεται CIN. Έτσι, σήμερα τα δεδομένα συνηγορούν υπέρ της ύπαρξης τριών διαφορετικών “Volgegrams” διαφορετικής αρχής και με διαφορετικά κλινικο-παθολογιανατομικά πρότυπα:
- I. Το μονοπάτι των οδοντωτών (serrated) αδενωμάτων που χαρακτηρίζεται από CIMP, MSI και μεταλλαγές του BRAF
- II. Το μονοπάτι των σωληνωδών αδενωμάτων που χαρακτηρίζεται από CIN, γενετικές μεταλλαγές που ενεργοποιούν το Wnt μονοπάτι (μεταλλάξεις APC, β-catenin κ.λ.π.) και φέρει συχνά μεταλλάξεις των KRAS και TP53 και
- III. Το μονοπάτι των λαχνωτών αδενωμάτων που χαρακτηρίζεται από διαφορετικά πρότυπα CIMP και φέρει συχνά μεταλλάξεις του KRAS αλλά σχεδόν ποτέ CIN (18).
Παρ’ όλο που το αναθεωρημένο αυτό μοντέλο συνταιριάζει τα έως σήμερα υπάρχοντα δεδομένα για το γενετικό και μοριακό προφίλ του ορθοκολικού καρκίνου υπάρχουν ακόμη σημαντικά κενά στη γνώση μας για την ασθένεια όπως οι μηχανισμοί που είναι υπεύθυνοι για τη CIN ή τη CIMP ή τα γενετικά-μοριακά συμβάματα που είναι καίρια για την ανάπτυξη μετάστασης από ένα πρωτοπαθή όγκο. Επίσης, ένα από τα πλέον ενδιαφέροντα ερωτήματα που αναδεικνύονται είναι εάν αυτοί οι τρεις διακριτοί μηχανισμοί στοχεύουν στο ίδιο κύτταρο ή εάν πρόκειται για τρεις τελείως διαφορετικές νοσολογικές οντότητες, όπως π.χ. συμβαίνει με τις λευχαιμίες οι οποίες σήμερα ταξινομούνται ανάλογα με το «γενεσιουργό» κύτταρο της νεοπλασίας ή/και το στάδιο διαφοροποίησης του.
Η πιο σημαντική οπτική γωνία αυτού του μοντέλου είναι ότι διακρίνει νεοπλάσματα με διαφορετική γενετική προέλευση και φυσική ιστορία γεγονός που έχει επίπτωση στην χάραξη στρατηγικών πρόληψης, προληπτικού ελέγχου και διάγνωσης καθώς και θεραπείας. Από την πλευρά της πρόληψης, εάν οι διαφορετικοί τύποι ορθοκολικού καρκίνου ξεκινούν από διαφορετικά κύτταρα, είναι πιθανό ότι η γενετική προδιάθεση και η έκθεση σε καρκινογόνα να ενεργοποιούν με διαφορετικούς μηχανισμού. Αυτή η ετερογένεια της νόσο θα μπορούσε να εξηγήσει την αδυναμία αναπαραγωγής των ευρημάτων διαφόρων μελετών συσχετίσεων ανάμεσα στην έκθεση σε καρκινογόνα, τον τρόπο ζωής ή σε γενετικούς πολυμορφισμούς και στον κίνδυνο εμφάνισης ορθοκολικού καρκίνου. Επίσης, οι γεωγραφικές διαφορές ανάλογα με τον υπότυπο του ορθοκολικού καρκίνου μπορεί να εξηγήσει τη διακύμανση που παρατηρείται σε διαφορετικές χώρες (19). Εάν αυτό ισχύει, παρεμβάσεις με σκοπό την πρόληψη του ορθοκολικού καρκίνου μπορούν να έχουν ευνοϊκή επίπτωση στο ένα τύπο καρκίνου και όχι στον άλλο, κάτι που μπορεί να οδηγήσει σε ψευδώς-αρνητικά αποτελέσματα (λόγω υποαντιπροσώπευσης του συγκεκριμένου υπότυπο στο σύνολο των νεοπλασμάτων).
Από την πλευρά του προληπτικού ελέγχου είναι υπό εξέλιξη οι προσπάθειες για την ανάπτυξη μοριακών δεικτών, για την έγκαιρη διάγνωση, στα κόπρανα και τον ορό των υγιών ατόμων. Αν η ασθένεια έχει τη γενετική ετερογένεια την οποία υποστηρίζουν τα έως σήμερα δεδομένα, τότε απαιτείται ένας συνδυασμός δεικτών που να καλύπτει όλα τα μονοπάτια που οδηγούν στον ορθοκολικό καρκίνο για να επιτευχθεί η επιθυμητή ευαισθησία. Επίσης, οι μέχρι σήμερα οδηγίες για τον ενδοσκοπικό έλεγχο βασίζονται στα δεδομένα για το «κλασσικό μοντέλο του Vogelstein» στο οποίο ο χρόνος για τη δημιουργία καρκινώματος από αδένωμα υπερβαίνει κατά κανόνα τα 15 έτη (20). Αντιθέτως, στο μονοπάτι των οδοντωτών αδενωμάτων η εξέλιξη είναι κατά πολύ ταχύτερη (21) υποδεικνύοντας ότι απαιτούνται διαφορετικές στρατηγικές προληπτικού ελέγχου ανάλογα με το γενετικό υπότυπο της νόσου.
Αλλά η μεγάλη συνεισφορά της μελέτης των μονοπατιών που οδηγούν σε ορθοκολικό καρκίνο σχετίζεται με τη εκλογή της ορθής θεραπευτικής στρατηγικής. Παρ’ όλες τις προσπάθειες και τα ευρήματα της από 10-ετίες έρευνας συνεχίζουμε να σκεφτόμαστε και να θεραπεύουμε τον ορθοκολικό καρκίνο σαν ενιαία νόσο. Αυτό είναι το σημείο που απαιτείται ριζική αλλαγή ιδιαίτερα στην εποχή των θεραπειών «μοριακής στόχευσης» (22). Μόλις πρόσφατα, η ανάλυση της μετάλλαξης του KRAS (που παρατηρείται στο 40% των περιπτώσεων) αντιπροσωπεύει ένα παράδειγμα για την ανάπτυξη βιοδεικτών στην εποχή της «θεραπείας μοριακής στόχευσης». Όπως έχει πλέον αποδειχθεί οι ασθενείς των οποίων ο πρωτοπαθής όγκος φέρει μεταλλαγές του KRAS είναι ανθεκτικοί σε θεραπεία με μονοκλωνικά αντισώματα έναντι του EGFR (23-27), γεγονός που οδήγησε τον Ευρωπαϊκό οργανισμό φαρμάκων (ΕΜΕΑ) σε αλλαγή της ένδειξη για τη χρήση αυτών των φαρμάκων.
Reference List
- Issa JP. Colon Cancer: It’s CIN or CIMP. Clinical Cancer Research 2008; 14(19):5939-5940.
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature 1998; 396(6712):643-649.
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature 1998; 396(6712):643-649.
- Vogelstein B. Cancer. A deadly inheritance. Nature 1990; 348(6303):681-682.
- Jen J, Kim H, Piantadosi S et al. Allelic loss of chromosome 18q and prognosis in colorectal cancer. N Engl J Med 1994; 331(4):213-221.
- Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983; 301(5895):89-92.
- Georgiades IB, Curtis LJ, Morris RM, Bird CC, Wyllie AH. Heterogeneity studies identify a subset of sporadic colorectal cancers without evidence for chromosomal or microsatellite instability. Oncogene 1999; 18(56):7933-7940.
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999; 96(15):8681-8686.
- Toyota M, Issa JP. Epigenetic changes in solid and hematopoietic tumors. Semin Oncol 2005; 32(5):521-530.
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999; 96(15):8681-8686.
- Cheng YW, Pincas H, Bacolod MD et al. CpG island methylator phenotype associates with low-degree chromosomal abnormalities in colorectal cancer. Clin Cancer Res 2008; 14(19):6005-6013.
- Goel A, Nagasaka T, Arnold CN et al. The CpG island methylator phenotype and chromosomal instability are inversely correlated in sporadic colorectal cancer. Gastroenterology 2007; 132(1):127-138.
- Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer 2004; 4(12):988-993.
- Wood LD, Parsons DW, Jones S et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318(5853):1108-1113.
- Etienne-Grimaldi MC, Formento JL, Francoual M et al. K-Ras mutations and treatment outcome in colorectal cancer patients receiving exclusive fluoropyrimidine therapy. Clin Cancer Res 2008; 14(15):4830-4835.
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10(8):789-799.
- Cheng YW, Pincas H, Bacolod MD et al. CpG island methylator phenotype associates with low-degree chromosomal abnormalities in colorectal cancer. Clin Cancer Res 2008; 14(19):6005-6013.
- Issa JP. Colon Cancer: It’s CIN or CIMP. Clinical Cancer Research 2008; 14(19):5939-5940.
- Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature 2004; 432(7015):338-341.
- Jones S, Chen WD, Parmigiani G et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A 2008; 105(11):4283-4288.
- Boparai KS, Mathus-Vliegen EM, Koornstra JJ et al. Increased colorectal cancer risk during follow-up in patients with hyperplastic polyposis syndrome: a multicentre cohort study. Gut 2009.
- Hamilton SR. Targeted therapy of cancer: new roles for pathologists in colorectal cancer. Mod Pathol 2008; 21 Suppl 2:S23-S30.
- Amado RG, Wolf M, Peeters M et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26(10):1626-1634.
- Bokemeyer C, Bondarenko I, Makhson A et al. Fluorouracil, Leucovorin, and Oxaliplatin With and Without Cetuximab in the First-Line Treatment of Metastatic Colorectal Cancer. J Clin Oncol 2008.
- Karapetis CS, Khambata-Ford S, Jonker DJ et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359(17):1757-1765.
- Souglakos J, Philips J, Wang R et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer 2009; 101(3):465-472.
- Van Cutsem E, Lang I, D’haens I et al. KRAS status and efficacy in the first-line treatment of patients with metastatic colorectal cancer (mCRC) treated with FOLFIRI with or without cetuximab: The CRYSTAL experience. J Clin Oncol 26[20s]. 2009.
Ref Type: Abstract

